

About Author:
Mahek Goel
Shri Baba Mastnath Institute of Pharmaceutical Science & Research
Asthal Bohar, Rohtak, Haryana (124001)
mahekgoel10@gmail.com
ABSTRACT
Parenteral drug delivery systems are the preparations that are given other than oral route. (Para-outside, enteric–intestine). Parenteral drug delivery systems are most preferred drug delivery systems as they meet many benefits over other dosage forms in many cases such as unconsciousness, nausea, in emergency clinical episodes. The Parenteral administration route is the most common and efficient for delivery of active drug substances with poor bio-availability and the drugs with a narrow therapeutic index. But parenteral route offers rapid onset of action with rapid declines of systemic drug level. For the sake of effective treatment it is often desirable to maintain systemic drug levels within the therapeutically effective concentration range for as long as treatment calls for. It requires frequent injection, which ultimately leads to patient discomfort. For this reason, drug delivery system which can reduce total number of injection throughout the effective treatment, improve patient compliance as well as pharmacoeconomic. These biodegradable injectable drug delivery system offer attractive opportunities for protein delivery and could possibly extend patent life of protein drugs.Parenteral drug delivery system seeks to optimize therapeutic index by providing immediate drug to the systemic pool in required quantity to treat– cardiac attacks, respiratory attacks. This article explores various prolonged release parenteral drug delivery system and their strategies of preparation, their potential benefits/drawbacks and in-vitro testing methods.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-1477
Introduction
The Parenteral administration route is the most effective and common form of delivery for active drug substances with metabolic bio-availabilities drug for which the bio-availability in limited by high first pass metabolism effect of other physicochemical limitation and for drugs with a narrow therapeutic index.
For this reason, whatever drug delivery technology that can reduce the total number of injection throughout the drug therapy period will be truly advantageous not only in terms of compliance, but also for potential to improve the quality of the therapy. Such reduction in frequency of drug dosing is achieved, in practice, by the use of specific formulation technologies that guarantee that the release of the active drug substance happens in a slow and predictable manner.
For several drugs, depending on the dose, it may be possible to reduce the injection frequency from daily to once or twice monthly or even less frequently. In addition to improving patient comfort, less frequent injection of drugs in the form of depot formulation smoothes out the plasma concentration time profiles by eliminating the peaks and valleys. Such smoothing out of the plasma profiles has the potential to not only boost the therapeutic benefit but also to reduce unwanted events and side effects.
The release can either be continuous or pulsatile depending on the structure of the device and the polymer characteristics, continuous release profiles are suitable to generate on ‘infusion like’ plasma level time profile in the systemic circulation without the necessity of hospitalization.
[adsense:468x15:2204050025]
Properties of ideal parenteral controlled drug delivery system:
• Safe from accidental release
• Simple to administer and remove
• Inert
• Biocompatible
• Mechanically strong
• Comfortable for the patient
• Capable of achieving high drug loading
• Readily processable
• Easy to fabricate and sterilize
• Free of leachable impurities
Advantage and Disadvantage:
1.Advantage:
(a) Convenience
(b) Compliance Potential for controlled release
(c) Avoiding the peak (risk of toxicity) at troughs (risk of ineffectiveness of conventional therapy)
(i) Reducing the dosing frequency.
(ii) Increasing patient compliance.
(d) Improved drug delivery
(e) Flexibility
2. Disadvantage:
(a) Invasive
(b) Danger of device failure
(c) Limited to potent drug
(d) Commercial disadvantage
Approaches used for the development of Parenteral controlled release formulations:
• Use of viscous, water miscible vehicles – aq. Solution of gelatin.
• Use of water immiscible vehicles – vegetable oils + aluminium monosterate
• Formation of thixotropic suspension
• Preparation of water insoluble drug derivatives – salts, complexes and esters.
• Dispersion in polymeric microspheres and microcapsules like lactide–glycolide homopolymers/ co-polymers.
• Co-administration of vasoconstrictors.
Polymers used in Parenteral controlled drug delivery system:
Generally,Biodegradable polymers are used for the preparation of parenteral controlled drug delivery system as it get degraded in the body and hence doesnot require removal from the body.
Classification of Biodegradable Polymers Biodegradable polymer may be classified based on the mechanism of release of the drug entrapped in it:
Natural - albumin starch, dextran, gelatin, fibrinogen, hemoglobin.
Synthetic - -cynoacrylates), poly ethyl--poly (alkyl cynoacrylates, poly amides. Nylon 6-10 nylon-cynoacrylates, poly butyl - 6-6, poly acryl amides, poly amino acid, poly urethane. Aliphatic poly esters are poly (lactic acid) poly lactide - co glycolide) poly glycolic acid, poly caprolactone, polydihydroxy butyrate, poly hydroxy butyrate co-valently cross linked protein, hydrogel.
Biodegradable polymers investigated for controlled drug delivery are Poly lactide / poly glycolide polymers, Poly anhydrides, Poly caprolactone, Poly orthoesters, Psuedo polyamino acid, Poly phosphazenes, Natural polymers
Desirable characteristics of an ideal Parenteral drug carrier
- Versatility in that carrier can deliver a variety of agents.
- High capacity to carry a sufficient quantity of drug per unit carrier to release therapeutic concentration to the target site without excessively loading host with the carrier.
- Restricting drug distribution to the desired target tissue.
- Uniform distribution within the capillary vasculature of the target tissue.
- Affording drug ready access to the parenchyma of target tissue.
- Restricting drug activity at the target site over a prolonged period.
- Minimizing systemic drug release during intravascular transit.
- Protecting drug from inactivation by plasma enzymes.
- Being biocompatible and minimally antigenic.
- Undergoing biologic degradation with prompt elimination and minimal toxicity of the breakdown products.
Major routes of Parenteral administration
- Intravenous (into a vein), e.g. many drugs, total parenteral nutrition (TPN)
- Intraarterial (into an artery), e.g. vasodilator drugs in the treatment of vasospasm and thrombolytic drugs for treatment of embolism
- Intramuscular (into a muscle), e.g. many vaccines, antibiotics, and long-acting psychoactive agents
- Intracardiac (into the heart), e.g. adrenalin during cardiopulmonary resuscitation (not commonly performed anymore)
- Subcutaneous (under the skin), e.g. insulin
- Intraosseous infusion (into the bone marrow) is, in effect, an indirect intravenous access because the bone marrow drains directly into the venous system. This route is occasionally used for drugs and fluids in emergency medicine and pediatrics when intravenous access is difficult
- Intradermal, (into the skin itself) is used for skin testing some allergens, and also for tattoos
- Intraperitoneal, (into the peritoneum) is predominantly used in veterinary medicine and animal testing for the administration of systemic drugs and fluids due to the ease of administration compared with other parenteral methods.
- Epidural (synonym: peridural) (injection or infusion into the epidural space), e.g. epidural anesthesia
- Intrathecal (injection or infusion into the cerebrospinal fluid), e.g. antibiotics, spinal anesthesia.
Biopharmaceutic considerations
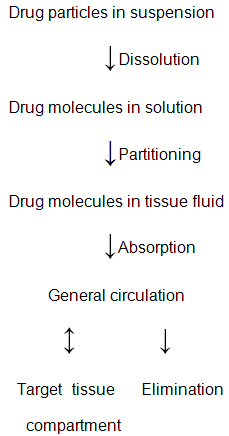
• For a drug administered intramuscularly or subcutaneously to reach the site of action to execute therapeutic activity, it must first be released from its formulation, transported from the injection site into the systemic circulation, and then delivered to the target tissue.
• For a drug administered parenteral in the form of a suspension in either aqueous or oleaginous vehicle the rate of delivery and the extent of availability of the drug to the site of action is frequently found to be controlled by the slowest (rate- limiting) step in the pharmacokinetic sequence as shown below.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
1.Effect of physiochemical properties-
1. Presence of other ingredients in the formulation and their interaction.
2. Solubility of drug in biological fluids.
3. Tissue fluid or vehicle partition coefficient of the drug.
4. Lipophilicity of the drug.
5. pKa value of the drug. pH value of the formulation.
6. Particle size and crystalline habit of drug solids.
7. Rate of dissolution of drug solids in the formulation vehicle.
All these parameters may play an important role in determining the time of onset, the intensity, and the duration of a therapeutic response of a drug delivered by an injectable controlled release or sustained formulation.
?The effect of the particle size on the dissolution of drug solids can also be applied to prolong the release of a drug. This can be achieved by increasing the particle size of drug solids in the formulation (macro crystal principle). It has proved that larger the particle size, more sustained is the serum drug level and the longer the duration of biological activity. Altering the solubility of the formulation can increase or decrease the rate of dissolution. E.g.Polymorphism. The polymorph with greater solubility has a faster rate of dissolution and therefore a more rapid absorption. Examples are Chloramphenicol and Novobiocin.
?For weakly acid or weakly basic drugs, drug molecules can exist in either an unionized or an ionized state, and the degree of ionization depends upon the dissociation constant Ka of the drug and the pH of the medium. As per Henderson-Hasselbalch equation, for acid drugs in medium with pH values below the pKa the drug molecules would exist predominantly as unionized form (absorbable form). On the other hand, the basic drugs exist in ionized form. The solubility of these drugs can be altered by changing the pH of the vehicle, which leads to an increase or decrease in the rate of dissolution in the hydrodynamic diffusion layer and the extent of partitioning of the drug molecules from the formulation to the tissue fluids at the injection site.
?The viscosity of the suspension can also affect the rate of dissolution by altering the solution diffusivity of drug molecule in the vehicle. For example, the use of an aq. solution having 35 %( v/v) glycerin as the vehicle decreased the acute subcutaneous toxicity of isoniazid and streptomycin sulphate.Certain suspensions show thixotropic behaviour i.e. when a suspension is stirred very gently or left standing for sometime, they show a nearly infinite viscosity, but they become fluidier in their consistency and flow more readily when shaken. The thixotropic suspension has three advantages-
Once the suspension reaches the injection site in the muscle tissue the suspension structure regenerates and a compact depot results.When the suspension is shaken before injection it becomes fluid enough to pass through hypodermic needle; In storage the suspension is stabilized by its structure and high viscosity.
2. Effect of physiological conditions
An increase in the muscular activity which produces an increase in blood flow to the muscles may yield an enhancement in the rate of drug absorption from the injection site. The criteria used to determine the route and the site parenteral drug administrations are as follows-
1. Desired rate and extent of systemic absorption
2. Total volume of the formulation to be administered
3. Dosing frequency
4. Inherent irritation, acidity/basicity, and/or concentration
5. Extent of local tissue irritation, nerve damage , and inadvertent blood vessel entry
6. Age and physical condition of the patient
In addition with the physicochemical properties such as blood flow from the injection site, and the physiological condition can also be relatively important for determining the onset and intensity of drug activity. The drug absorption can be enhanced by incorporating the hyaluronidase; because of it nature of spreading of injected drug solution over a large surface area of connective tissue that leads to the exposure of the drug molecules to a greater surface area for absorption.
Parenteral Depot System:
Depot: Long acting parenteral drug formulation are designed, ideally to provide slow constant, sustained, prolonged action.
Approaches used in Depot formulation:
1.Use of low aqueous soluble salt
2.Use of largest particle with crystalinity.
3.The suspension of the drug particle in vegetable oil and especially of gels with substances such as aluminum monasteries produces prolonged absorption rates.
Mechanism of controlled drug release based Depot formulations:
On the basis of different mechanism, depot formulation categories into four types
1) Dissolution controlled depot formulation
2) Adsorption type depot formulation
3)Encapsulation type depot formulation
4) Esterification type depot formulation
Dissolution-controlled depot formulations:
In this depot formulation the rate limiting step of drug absorption is the dissolution of drug particles in the formulation or in the tissue fluid surrounding the drug formulation. So drug absorption can control by slow dissolution of drug particle. The rate of drug dissolution (Q/t)d under sink conditions is defined by
(Q/t)d = Sa Ds Cs/hd (I)
Where Sa is the surface area of the drug particles in contact with the medium; Ds is the diffusion coefficient of drug molecules in the medium; Cs is the saturation solubility of drug in the medium; and hd is the thickness of the hydrodynamic diffusion layer surrounding each of the drug particle.
Basically, two approaches can be utilized to control the dissolution of drug particle to prolong the absorption and hence the therapeutic activity of the drug.
i) Formation of salt or complexes with low aqueous solubility. Typical examples are preparations of penicillin G procaine (Cs = 4 mg/ml) and penicillin G benzathine(Cs = 0.2 mg/ml) from the highly water-soluble alkali salts of penicillin G and preparations of naloxone pamoate and naltrexone-Zn-tannate from the water-soluble hydrochloride salts of naloxone and naltrexone, respectively.
ii) Suspension of Macrocrystals. Macrocrystals (large crystals) are known to dissolve more slowly than Microrystals (small crystals). This is called the macrocrystal principle (from equation-I, surface area of drug particle is directly proportional to dissolution) and can be applied to control the rate of drug dissolution. Typical example is the aqueous suspension of testosterone isobutyrate for intramuscular administration.
Adsorption-type depot preparation:
This depot preparation is formed by the binding of drug molecules to adsorbents. In this case only the unbound, free species of the drug is available for absorption. As soon as the unbound drug molecules are absorbed a fraction of the bound drug molecules is released to maintain equilibrium. This depot preparation is exemplified by vaccine preparations in which the antigens are bound to highly dispersed aluminum hydroxide gel to sustain their release and hence prolong the duration of stimulation of antibody formation.
Encapsulation-type depot preparations:
This depot preparation is prepared by encapsulating drug solids within a permeation barrier or dispersing drug particles in a diffusion matrix. The release of drug molecule is controlled by the rate of permeation across the permeation barrier and the rate of biodegradation of the barrier macromolecules. Both permeation barrier and diffusion matrix are fabricated from biodegradable or bioabsorbable macromolecules, such as gelatin, dextran, polylacticacid, lactide-glycolide copolymers, phospholipids, and long-chain fatty acids and glycerides. Typical examples are naltrexone pamoate-releasing biodegradable microcapsule, liposomes, and norethindrone-releasing biodegradable lactide-glycolide copolymer beads.
Esterification-type depot preparations:
This depot preparation is produced by esterifying a drug to form a bioconvertible Prodrug-type ester and then formulating it in an Injectable formulation. This chemical approach depends upon number of enzyme (esterase) present at the injection site. This formulation forms a drug reservoir at the site of Injection. The rate of drug absorption is controlled by the interfacial partitioning of drug esters from the reservoir to the tissue fluid and the rate of bioconversion of drug esters to regenerate active drug molecules. It is exemplified by the fluphenazine enanthate, nandrolone decanoate in oleaginous solution.
Classification of parenteral controlled drug delivery system:
- Injectables
- Implants
- Infusion Devices
1.Injectables
Solid Lipid Nanoparticles
The concept of lipid nanoparticles for injectable delivery was developed from submicron sized parenteral fat o/w emulsion used for parenteral nutrition viz. Intralipid .This gave birth to the idea of encapsulating lipophilic drugs into oil droplets. The only drawback associated with these submicron emulsions was the low viscosity of the droplets, causing fast release and susceptibility of the incorporated actives towards degradation by the aqueous continuous phase.In 1990s, researchers (Mueller and coworkers and Gasco and coworkers) started exploring the potential of nanoparticles-based solid lipids or SLNs in the drug delivery. SLN are submicron colloidal particles composed of a biocompatible/biodegradable lipid matrix that is solid at body temperature and exhibit size range in between 100 and 400 nm.These are composed generally of physiological lipid dispersed in water or in aqueous surfactant solution.SLNs combine advantages of polymeric nanoparticles,fat emulsions and liposomes and are simultaneously capable of avoiding some of their disadvantages.The lipid nanopellets are prepared by first melting the lipid and then it was dispersed in a hot surfactant solution by stirring or ultrasonic treatment.The hot microemulsion containing the lipid was poured into cold water leading to solidification of nanoparticles.SLNs can also be prepared by another diverse homogenization method at higher pressure for either melted or solid lipid.Various drug molecules have been incorporated in injectable SLN for treatment of different diseases.
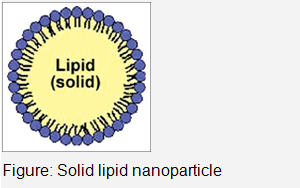
Advantages
- Particulate nature
- Amenability to encapsulate hydrophilic and hydrophobic drugs
- Ability to sustain the release of incorporated drug
- Ability to prevent chemical, photochemical, or oxidative degradation of drug
- Ability to immobilize drug in the solid matrix
- Ease of scale-up and manufacture
- Low cost of solid lipids as compared with phospholipids and biodegradable polymers
Nanodispersions
There are broader applications of lipid systems in parenteral drug delivery. However, with specific new chemical entities, it has been limited due to the following reasons:
- Only a small number of parenteral lipid excipients are approved.
- There is increasing number of drugs that are partially or not soluble in conventional oils and other lipid solvents.
-
The ongoing requirement for site-specific targeting and controlled drug release. These advanced parenteral lipid nano dispersions include nanoemulsions and nanosuspensions.
Characteristics of desired parenteral formulation of a marketed drug - Exhibits high drug solubilization/loading capacity.
- Is at least as efficacious as the marketed formulation.
- It has a shelf-life of at least 2 years.
- Should be easily processed and manufactured using standard equipment.
- Is amenable to fast track development status by addressing 'unmet medical needs.
Nanoemulsions
Nanoemulsions or miniemulsions are transparent or translucent oil-in-water (o/w) or water-in-oil droplets with a mean droplet diameter in the range between 100 and 500 nm.They are also known as submicron emulsions and unlike the thermodynamically stable microemulsions, nanoemulsions are kinetically stable with great stability in suspension due to their small droplet size.
These differs from each other in certain aspects:
Nanoemulsions:Knetically stable,contains less amount of surfactant and can be formed only after application of high shear.
Microemulsion:Thermodynamically stable,contains more amount of surfactant and can be formed spontaneously.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Nanosuspensions/Nanosuspensions
Nanosuspensions of drugs are submicron colloidal dispersions of drug particles which are stabilized by surfactants. Nanocrystals are crystals of poorly water soluble drug in nanosize which when dispersed in water produce nnocrystals.To produce nanosuspensions,the drug powder is dispersed in an aqueous surfactant solution by high speed stirring which can last from hours upto several days.The obtained macrosuspension is passed through a high-speed homogenizer that generates nanosuspension. Nanosuspensions so obtained have exceptionally high interfacial energy.
Advantages of nanosuspensions
Used to formulate drugs that are insoluble in both water and oil, and are usually employed when the use of other lipid-based systems are limited.Nanocrystals have an advantage of higher loading (pto 90% of crystalline particle is drug).In the case of high melting point compounds, solubilization in any solvent is difficult; nanosuspensions can be used to maintain these drugs in a preferred crystalline state of sufficiently small size for intravenous administration.
Niosomes
Niosomes are nonionic surfactant vesicles obtained on hydration of synthetic nonionic surfactants of the alkyl or dialkyl polyglycerol ether class, with or without incorporation of cholesterol or other lipids.These are bilayered structure which can entrap both hydrophilic and lipophilic drugs either in an aqueous layer or in vesicular membrane,made up of lipids.Various drugs incorporated into niosomes by different methods are shown as below:
|
METHOD OF PREPARATION |
DRUGS INCORPORATED |
|
Ether injection Hand shaking Sonication |
Doxorubicin Methotrexate Vasopressin |
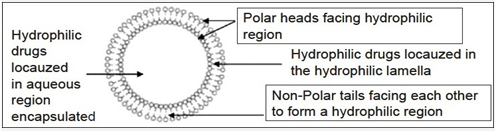
Figure 3 :Niosome structure
Advantages of niosomes
- They entrap solute in a manner analogous to liposomes.
- They are osmotically active and stable.
- Handling and storage of surfactants requires no special conditions.
- They possess an infrastructure consisting of hydrophobic and hydrophilic moieties together and as a result can accommodate drug molecules with a wide range of solubilities.
- They exhibit flexibility in their structural characteristics (composition, fluidity, and size) and can be designed according to desired application.
- They improve oral bioavailability of poorly absorbed drugs and enhance skin penetration of drugs.
- They allow their surface for attachment of hydrophilic group and can incorporate hydrophilic moieties in bilayer to bring about changes in their in vivo behavior.
- The surfactants are biodegradable, biocompatible, and nonimmunogenic.
- They improve the therapeutic performance of the drug molecules by delaying the clearance from the circulation, protecting the drug from biological environment, and restricting effects to target cells.
- Niosomal dispersion in an aqueous phase can be emulsified in a nonaqueous phase to regulate the delivery rate of drug and administer normal vesicle in external nonaqueous phase.
Liposomes
Liposomes are formed by the self-assembly of phospholipid molecules in an aqueous environment. The amphiphilic phospholipid molecules form a closed bilayer sphere in an attempt to shield their hydrophobic groups from the aqueous environment while still maintaining contact with the aqueous phase via the hydrophilic head group. When suitably dispersed they consist of a series of concentric bilayers alternating with aqueous compartments. Water or lipid soluble substances can be entrapped within their aqueous or lipid phase respectively. Depending on the phospholipids used and the ionic composition of the medium,liposomes of various sizes and shapes can be obtained.Furthermore,antibodies can be covalently coupled to liposomes to enhance their cell specificity.
Microspheres
Microsheres are solid,spherical particles containing dispersed molecules either in solution or in crystalline form.Numerous biodegradable polymers have been investigated for preparation of microspheres as depot formulation.Since drug release from this system is rate limited by dissolution of the matrix,so it is find that small polymer matrices released drug at a faster rate than larger ones.The application of biodegradable microspheres to deliver small molecules, proteins, and macromolecules using multiple routes of administration has been widely investigated and several products have been brought to market in the last 10–20 years.The method of preparation consists of suspending the drug in a biodegradable polymer,followed by reducing the mixture to particles of the order of 600 μm,which are then injected as a suspension in carboxymethyl cellulose solution. For peptide or protein containing microspheres mainly three processes were studied more intensively, namely the w/o/w –technique phase separation methods and to some extent spray drying and by w/o/w emulsion-solvent evaporation method. The microsphere release drug in a zero order fashion over 1 to 3 months after intramuscular or subcutaneous injection into animals. PLGA microsphere had been also used for delivery of glycoprotein (GP) IIb/IIIa antagonist, plasmid DNA, Interleukin-1α and prolidase enzyme.
To prolong the circulation, nanoparticles should be small enough.By taking an advantage of their rapid uptake by the RES and sequestration by liver kuffer’s cells. The RES consist of phagocytic cells designed to cleanse unwanted cell debris and foreign particles E.g. Nanoparticles loaded with radioisotope technetium-99n can be used to image hepatic pathologies.
Magnetic microspheres
Magnetic microspheres were developed to minimize reticuloendothelial clearance and to increase target site specificity.They can be used to entrap a wide variety of drugs .Magnetic microspheres can be prepared from albumin and magnetite. They are about 1.0μm in size,which is small enough to allow them to inject intravenously. These are relatively non-toxic and non-reactive with blood components.
Typically,Magnetic microspheres are infused into an artery supplying a given target site.A magnet of sufficient field strength is then externally placed over the target area to localize microspheres at the capillary bedin this region.
Specialised Emulsions
Emulsions are dispersions of one liquid inside a second liquid, where liquid being immiscible, such dispersions are stabilized by emulsifiers which coat droplets and prevent droplets coalescence by reducing interfacial tension. The aq. insoluble drugs can be formulated by dissolving it in the oil phase of an emulsion and drugs that prone to hydrolysis can be prevented by means of emulsification technique.The emulsified drugs can be diverted from liver on i.v injection by incorporating polyoxyethylene surfactants as emulsifiers. Emulsion may be used to reduce toxicity. Eg. Use w/o emulsion of amphotericin deoxycholate reduces the incidence and severity of renal impairment.
Multiple Emulsions
In the multiple emulsions the dispersed phase contains smaller droplets that have same composition as the external phase-“double emulsion” (double emulsification) or “liquid membrane system” (the liquid film that separates the liquid phases and acts as a thin semi permeable film through which solute diffuse in order to transverse from one to another). They can be o/w/o or w/o/w type.Because of their vesicular nature, these multiple emulsion has immense potential as drug carrier as like liposomal vesicles.
Emulsomes
Emulosomes are lipid based drug delivery systems, which are poorly water soluble. Emulosome particles are basically consisting of microscopic lipid assembly with a polar core. These systems are prepared by melt expression or emulsion solvent diffusive extraction. In place of emulgents, phospholipids or used for stabilization which in turn leads to the formation of bilayered membrane similar to liposomes.Thus these systems combine the characteristics of lipid sphere (apolar core) and liposome (surface).
These can be safely used as parenterals administration of drug, as adjuvants for vaccines, as carrier container for targeted drug delivery to liver, brain and RES rich organs.
Resealed erythrocytes
Resealed erythrocytes act as promising drug carriers. In which the erythrocytes lose their haemoglobin content at large when they are processed in laboratory for transient permeation by applying various methods that includes osmolysis, electrical breakdown/electro-encapsulation, endocytosis and other means and modes of perturbation. The haemoglobin loss provides intracellular space for drug to be incorporated.
Drug transient permeation phase - an equilibrium is established between intra and extra cellular contents and there after , the permeable erythrocytes membrane can be resealed by restoring tonicity of the media and incubating them at 37°C. The biological half life of resealed erythrocytes is 60-120 days depending on the method of drug loading chosen. They are biodegradable, biocompatible, and non-immunogenic and can be used as circulatory drug depots.The contents of the erythrocytes are released following phagocytosis /diffusion through the membrane/ some other transport mechanism.
Cyclodextrins(Amphiphilic β-cyclodextrin)
Cyclodextrins are water soluble cyclic carbohydrate compounds with hydrophobic cavity. These compounds can form inclusion complexes with hydrophobic guest molecules endowing such molecules with aq. solubility. CD’s are used parenterally chiefly as pharmaceutical solubilizing agents
Only modified forms-
Hydroxypropyl β-cyclodextrin Parenteral molecular carriers Sulphobutyl β-cyclodextrin
Nimodipine aq. suspension –the relative solubility of the drug is increased with no change in drug AUC. Propofol (inclusion complex) - decreases the drug sleep induction time when compared with administration as o/w emulsion.
Prodrugs
The basic prodrug concept deals with chemical modification of the drugs following the parenteral administration undergo suitable changes yielding active principles at the site/body compartment exempting other body parts from unnecessary exposure to drugs. The organs which are targeted with active drugs contain the enzyme responsible for the metabolism of prodrug.
These drugs are so designed that prodrug and regenerated metabolites both are soft to the biological system being isoelectronic/isosteric with drug metabolic analogues; they possess better pharmacodynamic activity and site specificity.
Aquasomes
Aquasomes are 3-layered self assembling composition with –
As these are solid or glassy particles dispersed in an aq. environment, they exhibit the physical properties of colloids and their mechanism of action is controlled by their surface chemistry.Ceramic carbon nano-crystalline particle core coated with glassy cellobiose (or) degradable calcium phosphate monomer crystalline particle core coated with glassy pyridoxal-5-phosphate. Subsequently, drug /enzyme is non-covalently bond to the outer coating.
Aquasomes deliver their contents through a combination of specific targeting, molecular shielding and a slow sustained release process. Aquasome technology represents a platform system for preserving conformational integrity and biochemical stability of bioactives. For gene therapy, a 5- layered composition of Aquasome comprised of the ceramic mono crystalline core, the polyhydroxyl oligomeric film coating, the noncovalently bound layer of therapeutic gene segment, an additional conserve viral membrane proteins have been proposed for gene therapy.
Dendrimers
The term “Dendrimer” is derived from Greek words - Dendron means type & meros means part. Dendrimers are highly branched 3-D-macromolecules with highly controlled structures with all bounds emanating from a central core. Dendrimer construction methods are fundamentally divided into two- 1. Divergent method- In this method, one branching unit after another is successfully attached to the core molecule. Hence, the multiplication of the number of the peripheral groups is dependent on the branching multiplicity. 2. Convergent method- involves the opposite course, where the skeleton in built stepwise starting from the end group towards the inside and finally treated with a core molecule to produce the Dendrimer. The internal structures acquire a star like appearance and the final products as appearance called “Starburst Dendrimers”.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
2.Implant system
Implant systems are indicated incase of chronic therapy, such as hormone replacement therapy and chemical castration in the treatment of prostrate cancer. Parenteral implants can be highly viscous liquids or semisolid formulations both of which may be injected with a needle and implants may be in the form of tiny rods impregnated with a drug substance/ a liquid which gel following administration.
These parenteral implants are prepared from polymeric materials including – polysaccharides, polylactic acid coglycolic acid (no need for surgical removal of the implant after treatment) and non degradable methacrylates. Various principles/mechanisms such as diffusion, dissolution, vapour–pressure, osmosis, and ion–exchange etc., are exploited for implant system.
Implants can be further classified as:
1.Solid implants
Solid implants typically exhibit biphasic release kinetics, with initial burst of drug is usually due to the release of drug deposited on the surface of the implant although zero order kinetics may be achieved by. Eg. Coating the implant drug impermeable material.
Overall drug release may be controlled by varying polymer composition- an increase in the level of lactic acid in a polylactic acid co-glycolic acid copolymer retards drug release and increase in polymer molecular weight also retards drug release and prolongs drug effects. The conventional pulsatile delivery systems such as electrically triggered release of insulin from polydimethylaminopropylacrylamide gels. A viscous gelatin solution or galactoxyloglucan gel of mitomycin C administered intra peritorially prolongs potentials and plasma clearance.
In order to achieve high drug doses in traditionally inaccessible areas such as CNS, bone tissue and beyond the blood –retinal barrier implants could effectively be used clinically. Ethylene vinyl acetate co polymer dexamethasone intracranial base implants produce high drug levels in brain with plasma levels remain normal. Additionally polylactic acid co-glycolic acid sclera implants containing gancyclovir for the treatment of cytomegalovirus infection could maintain effective therapeutic levels of the drug in vitreous humour and retina/choroid for over a period of 3-5 months. It is concluded that implant systems offer a means of achieving high drug concentration in areas that are usually inaccessible to peripherally or vascularly administered drug. In addition, the high drug levels are maintained in sustained manner in these areas.
2.In-Situ forming implants
Biodegradable injectable in situ forming drug delivery systems represent an attractive alternative to microspheres and implants as parenteral depot systems. The controlled release of bioactive macromolecules via (semi-) solid in situ forming systems has a number of advantages, such as:
- ease of administration,
- less complicated fabrication,
- less stressful manufacturing conditions for sensitive drug molecules.
From a manufacturing point of view, in situ forming depot systems offer the advantage that they are relatively simple to manufacture from polymers adapted for this approach. Compared with microspheres, which have to be washed and isolated after preparation, operating expenses for the production of in situ forming applications are marginal, thus lowering investment and manufacturing costs.
Classification of injectable in situ forming implants (according to their mechanism of depot formation)
Thermoplastic pastes Semi-solid polymers can be injected when melted and form a depot upon cooling to body temperature. The requirements for such In Situ Forming Devices (ISFD) include low melting or glass transition temperatures in the range of 25 to 65°C and an intrinsic viscosity in the range of 0.05 to 0.8 dl/g. Below the viscosity of 0.05 dl/g, no delayed release could be observed, where as above 0.8 dl/g the ISFD was no longer injectable using a needle. At injection temperature above 37?C but below 65?C these polymers behave like viscous fluids which solidify to highly viscous depots. Drugs are incorporated into the molten polymer by mixing without the application of solvents. Thermoplastic pastes allow local drug delivery at sites of surgical interventions for the delivery of antibiotic or cytotoxic agents. Alternatively, they can be used to generate a subcutaneous drug reservoir from which diffusion occurs into the systemic circulation.
In situ cross-linked polymer systems
The formation of a cross-linked polymer network is advantageous because of the possibility to control the diffusion of hydrophilic macromolecules. Such a system could ideally release peptides and proteins over a prolonged period of time. Cross-linked polymer network can be found in situ by free radical reactions initiated by heat (thermosets) or absorption of photon or ionic interactions between small cation and polymer anions.It requires free radical producing agents such as benzoyl peroxide into the body which may induce tumor promotion.Only the calcium concentration in the eye led to in situ formation of alginate formulations.Despite these applications, there are two important factors which limit the use of calcium-alginate. The first factor is their potential immunogenicity and the second is longer time in vivo degradability. In situ cross-linking implants have been a challenging objective, as polymers containing double-bonds and free radical-initiation are necessary.
In situ polymer precipitation
A water-insoluble and biodegradable polymer is dissolved in a biocompatible organic solvent to which a drug is added, forming a solution or suspension after mixing. When this formulation is injected into the body, the water-miscible organic solvent dissipates and water penetrates into the organic phase. This leads to phase separation and precipitation of the polymer, forming a depot at the site of injection. This method has been developed by ARTIX Laboratories and is designated as the Atrigel technology , which used as a drug carrier for Eligard,contains the leuteinizing hormone releasing hormone (LHRH) agonist leuprolide acetate (7.5, 22.5 or 30mg) and poly(lactide-co-glycolic acid)(PLGA) 75/25 dissolved in N-methyl-2-pyrrolidone (NMP) in a 45:55 (m/m) polymer:NMP ratio. This system led to suppression of testosterone levels in dogs for approximately 91d. One of the problem with these system is the possibility of a burst in drug release especially during the first few hours after injection into the body. In order to control the burst effect, four factors have been examined, the concentration of polymer in the solvent, the molecular weight of the polymer, the solvent used and the addition of surfactant Also the drug burst is directly related to the dynamics of the phase inversion.
Thermally induced gelling systems
Numerous polymers show abrupt changes in solubility as a function of environmental temperature. The clear advantage is the ability to solubilize the water-insoluble drug substances, such as paclitaxel, which allows a prolonged release for more than 50 days. Sol-gel transitions occur around 308ºC at polymer concentrations of 15 to 23% (w/w) in aqueous solution. Biocompatibility and toxicity do not seem to be problematic. An aqueous solution of low molecular weight PEGPLGA-PEG (550-2810-550) triblock copolymers becomes gel at body temperature. Two model drugs, Ketoprofen (hydrophilic drug) and spirinolactone (hydrophobic drug) were released from the PEG-PLGA-PEG triblock copolymer hydrogel over 2 weeks with first order release profile and over 2 months with an s-shaped release profile, respectively. The higher the initial polymer solution concentration, the slower was the drug release rate observed,due to tighter polymer-polymer contacts among the gel at higher concentrations of the polymer
The release rate strongly depended on the liposome size and composition (i.e. addition of cholesterol), and on the presence of phospholipase in the release medium
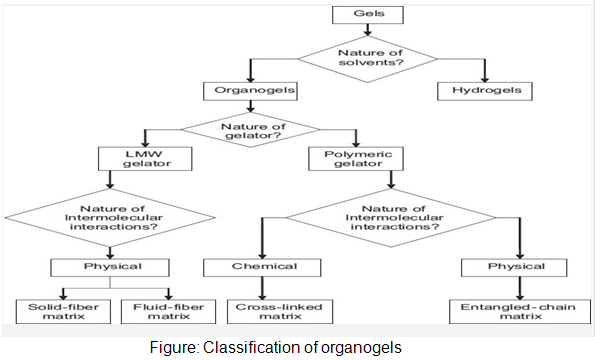
In-situ forming organogels
Organogels are semi-solid systems in which an organic liquid phase is immobilized by a three-dimensional network composed of self assembled, intertwined gelator fibers. Despite their majority liquid composition, these systems demonstrate the appearance and rheological behavior of solids. Organogels are composed of water insoluble amphiphilic lipids, which swell in water and forms various types of lyotropic liquid crystals. The amphiphilic lipids examined for drug delivery are glycerol monooleate, glycerol monopalmitostearate, glycerol monolinoleate, sorbitan monostearate (SMS) and different gelation modifiers (polysorbates 20 and 80) in various organic solvents and oils. These compound forms a cubic liquid crystal phase upon injection into an aqueous medium which is gel like and highly viscous.SMS organogels containing either w/o or vesicular in water in oil (v/w/o) emulsion.Intramuscular administration of the v/w/o gel yielded the long lasting depot effect (48hr).
Organogel formulation used in parenteral drug delivery
Rivastigmine and leuprolide are sed in organogel formulation by subcutaneous route, in which N-stearoyl l-alanine methyl or ethyl ester is used as organogelators.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org






