

 About Author: Rahul Sharma (M.Pharm - Pharmaceutics)
About Author: Rahul Sharma (M.Pharm - Pharmaceutics)
Lord Shiva College of Pharmacy, Sirsa, Haryana (India)
Abstract:
Bioanalysis, employed for the quantitative determination of drugs and their metabolites in biological fluids, plays a significant role in the evaluation and interpretation of bioequivalence, pharmacokinetic (PK), and toxicokinetic studies. The quality of these studies, which are often used to support regulatory filings, is directly related to the quality of the underlying bioanalytical data (1). It is essential to employ well-characterized and fully validated analytical methods to yield reliable results which can be satisfactorily interpreted as well as to emphasize that each analytical technique has its own characteristics, which will vary from analyte to analyte (2).
[adsense:336x280:8701650588]
Reference ID: PHARMATUTOR-ART-1132
Introduction
The development of the sound bioanalytical method is of paramount importance during the process of drug discovery and development culminating in marketing approval (4). It is a set of various procedures, mainly used for the determination of drugs and their metabolites in biological matrices such as urine, plasma, and serum.

Method development starts with literature survey of the molecule in which we find the nature of the molecule its pKa, solubility, molecular weight etc. Accordingly we select the appropriate chromatographic technique to determine the initial conditions of the separation. Methods for analyzing drugs by HPLC and LCMS-MS can be developed, provided one has knowledge about the nature of the sample, namely, its molecular weight, polarity, ionic character, pKa values and the solubility parameter. Method development cannot be standardized across the board because method development is unique and specific for each drug candidate. It also depends on the nature of the sample, sensitivity required etc (3,5).
[adsense:468x15:2204050025]
Steps and Considerations for Method Development
Before beginning method development process, we need to review certain parameters and continue accordingly, inorder to take those parameters under consideration for an effective and suitable method for a particular analyte (6). The various steps and procedures which plays a vital role in the method development process of an analyte are:
a.) Information about the sample
The knowledge about the sample can provide valuable clues for the best choice of initial conditions for a chromatographic separation. Depending on the use made of this sample information, different approaches to LCMS method development are possible. The necessary information about an analyte before beginning the method development process are (6):
· Chemical structure (functionality) of compounds
· Molecular weight
· pKa value
· Sample Solubility and stability
· Toxicity, purity
· Concentration range of compounds in samples of interest
· Metabolites
b.) Chromatographic Approaches
By a valuable collection of information about the sample, we can easily select the chromatography method for the analyte. Different approaches are:
· First Reversed phase chromatography should be tried.
· Than normal phase chromatography should be tried if reverse phase fails.
· Ion pair chromatography is tried for ion forming compounds but before going for ion pairing, ion suppression by pH control reverse phase chromatography should be tried should be tried (3,6,7).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
c.) Sample Preparation Approaches
Sample preparation is an essential part of high importance when an LC-MS/MS method is developed for the bioanalysis. Because of the large amount of proteins in plasma samples, conventional HPLC column will not tolerate the direct introduction of plasma, therefore most bioanalytical process have a sample preparation stage (3,8).
The main aim of sample preparation step is to provide a reproducible and homogenous solution suitable for injection into column. The main characteristics of the sample which can be used after sample preparation step for injection into the column are:
1. It should be free from possible interferences.
2. It should not damage the column.
3. Compatible with proposed LCMS method.
4. Solvent used for preparation of the sample should be compatible with mobile phase and should not significantly affect the retention and resolution of the analyte (3).
The various types of sample preparation approaches are:
· Solid-phase extraction
· Liquid-Liquid extraction
· Protein Precipitation
· Filtration
· Dilution
· Homogenization
· Ultrafiltration
· Lyophilization
· Vortexing
· Distillation
· Turbulent flow chromatography
Other important reason to have a sample preparation step include reducing the matrix components and also elimination of ion suppression. From all the above given sample preparation approaches, Solid phase extraction, Liquid liquid extraction and protein precipitation are mainly used for bioanalytical method development process (3,8).
(a) Solid-phase Extraction
Solid-phase extraction (SPE) is a widely used sample preparation technique for the isolation of selected analytes, usually from the mobile phase. The analytes are transferred to the solid (stationary phase) where they are retained for the duration of sampling process, than the analytes recovered by using reconstitution solution (9). Solid phase extraction stationary phase includes silica, highly cross linked polystyrene-divinylbenzene copolymer, carbon and other oxide materials such as alumina, magnesia, and zirconia (10).Disposable cartridges containing silica-based chemically bonded sorbents of a suitable size is used in SPE (9).
SPE cartridges:SPE cartridges are designed to simply and improve sample preparation by combining the right sorbent chemistry and methodology. There are five available Oasis sorbent chemistries which are designed to meet of all sample preparation needs. They are all built upon a unique, water-wettable Oasis HLB (Hydrophilic-Lipophilic Balance) copolymer and provide exceptional results. The sulfonic acid MCX (Mixed-Mode Cation-exchange), and quaternary amine MAX (Mixed-Mode Anion-exchange) derivatives of Oasis HLB provide dual modes of retention (e.g. both reversed- phase and ion exchange retention modes available), enabling greater cleanup, selectivity and sensitivity for both acid and/or basic compounds- even if the sorbent in the wells run dry.
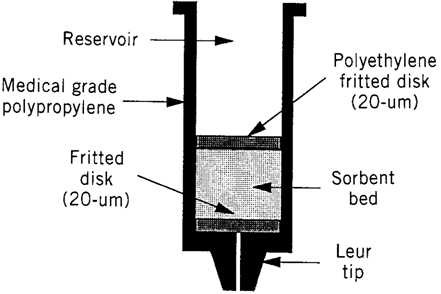
The carboxylic acid WCX (Weak Cation-exchange) and the piperazine WAX (Weak Anion-exchange) are also derivatives of Oasis HLB and provide dual modes of retention. These sorbents are specifically designed to offer the same benefits and features as HLB with ability to retain and release strong bases (e.g. quaternary amines) and strong acids (e.g. sulfonates) respectively. All of the five patented Oasis chemistries are available in several device formats (e.g. cartridges, 96-well plates, and melution plates (3,9,10).
(b) Liquid-liquid Extraction (LLE)
Liquid-liquid extraction is useful for separating analytes from interferences by partitioning the sample between two immiscible liquids or phases. One phase in LLE often is aqueous and second phase an organic solvent. More hydrophilic compounds prefer the polar aqueous phase, where as more hydrophobic compounds will be found mainly in the organic solvents. Analyte extracted into the organic phase are easily recovered by evaporation of the solvent, while analytes extracted in to the aqueous phase can often be injected directly on to a reversed-phase column. The technique is simple, rapid and has relatively small cost factor per sample when compared to others. The extraction containing drug can be evaporated to dryness and the residue reconstituted in a smaller volume of a appropriate solvent (preferably mobile phase). Near quantitative recoveries (90%) of most drugs can be obtained through multiple continuous extraction (3,5).Problems associated with LLE are (3):
· Formation of emulsion
· Adsorption of analyte to matrix / excipients
· Binding of analyte to high molecular weight excipients
· Mutual partial solubility of two phases employed for LLE (chloroform-water)
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
(c) Protein Precipitation
Protein precipitation is the simple method of extraction as compared to the LLE and SPE. This can be carried out by using the suitable organic solvents which has good solubility of the analyte and protein precipitating properties. Acetonitrile is the first choice of solvent for protein precipitation due to its complete precipitation of proteins and methanol is the second choice. In this sample preparation approach mainly removal of protein by denaturation and precipitation is carried out. After protein precipitation the supernatant obtained can be injected directly in to the HPLC or it can be evaporated and reconstituted with the mobile phase and further clean up of the sample can be carried out by using micro centrifuge at very high speed (4,5,8).
Parameters Influencing LC-MS/MS Method Development
During the process of developing a method for the analysis of any analyte, there are some factors or we can say some parameters which greatly influences the whole process of method development. Due to this reason, we cannot neglect them and establish our method accordingly. These parameters are (3,5):
· Selection of Internal Standard
· Selection of Mobile phase
· Selection of Column
· Role of pH
· Role of Buffers
· Role of flow rate
· Role of Temperature
a.) Selection of Internal Standard
The best internal standard is an isotopically labelled version of the molecule you want to quantify. An isotopically labeled internal standard will have a similar extraction recovery, ionization response in ESI mass spectrometry, and a similar chromatographic retention time. Internal standard mainly added to a sample in known concentration to facilitate the qualitative indentification and/or quantitative determination of the sample components (3,5).
b.) Selection of Mobile Phase
A successful chromatographic separation depends upon differences in the interaction of the solutes with the mobile phase and the stationary phase, and in liquid chromatography, choice and variation of the mobile phase is of critical importance in achieving optimum efficiency (30). If the sample contains ionic or ionisable compounds, then use of a buffered mobile phase to ensure reproducible results. Under unfavorable circumstances, pH changes as little as 0.1 pH units can have a significant effect on the separation. For LCMS-MS ammonium acetate and formate are suitable for buffer mobile phase as they are volatile in nature; usually 2-10mM concentration is adequate but concentration upto 50mM can be used. Acetonitrile is the preferred organic modifier in reverse-phase chromatography (5).
c.) Selection of Column
The HPLC column is the heart of the method, critical performing the separation. The column must possess the selectivity, efficiency and reproducibility to provide good separation. Commonly used reversed phase columns are C18 (octadecyl silane,), C8 (octyl silane,) phenyl and cyano. They are chemically different bonded phases and demonstrate significant changes in the selectivity using the same mobile phase During method development selection of column can be streamlined by starting with shorter columns (150,100 or even 50mm long.). By selecting a shorter column with an appropriate phase run time can be minimized so that an elution order and an optimum mobile phase can be quickly determined. It is also advantageous to consider the column internal diameter, many laboratories use 4.6mm ID as standard, but it is worth considering use of 4mm ID column as an alternative. This requires only 75% of the solvent flow than that of 4.6mm column. Selecting an appropriate stationary phase can also help to improve the efficiency of method development. For example, a octyl phase (C8) can provide time saving over a octadecyl (C18) as it doesn’t retain analytes as strongly as the C18 phase (3,5).
d.) Role of pH
pH is another factor in the resolution that will affect the selectivity of the separation in reversed-phase HPLC. Selecting the proper buffer pH is necessary to reproducible separation of ionizable compounds by Reversed-Phase HPLC. Selecting an improper pH for ionizables analyte often leads to asymmetric peaks that are broad, tail, split, or shoulder. Sharp, symmetrical peaks are necessary in quantitative analysis in order to achieve low detection limits. When the pH = pKa for the analyte, it is half ionized, i.e. the concentrations of the ionized and unionized species are equal. As mostly all of the pH caused changes in the retention occur within ± 1.5 pH units of the pKa value, it is best to adjust the mobile phase to pH values at least ± 1.5 pH units of above or below the pKa to ensure practically 100% unionization of analyte for retention purposes. Generally at low pH peak tailing is minimized and method ruggedness is maximized. On the other hand, operating in the intermediate pH offers an advantage in increased analyte retention and selectivity (3,5).
e.) Role of Buffer
In Reverse-Phase liquid chromatography mobile phase pH values are usually between 2.0 and 7.5. Buffers are needed when an analyte is ionizable under Reverse-Phase conditions or the sample solution is outside this pH range. Analytes ionizable under Reverse-phase conditions often have amine or acid functional groups with pKa between 1.0 and 11.0. A correctly chosen buffer pH will ensure that the ionizable functional group is in a single form, whether ionic or neutral. If the sample solution is at pH damaging to the column, the buffer will quickly bring the pH of the injected solution to a less harmful pH. If the analyte contain only amine functional groups Buffer selection is easier. Most amines will be in cationic form at pH value less than 9.0, so any buffer effective at pH 7.0 or lower will work. Buffer at pH 7.0 are used, even though pH of water is 7.0, because amine retention and peak shapes are pH dependent. As pH is lowered amine retention time shortens and peak shapes sharpens as the buffer protonates the acidic silanols on the silica surface. Any buffer with a pKa less than 7.0 is suitable, but phosphate buffer of pH 3.0 is found to be best for amines (3,5).
f.) Role of Flow rate
Flow rate, more for isocratic than gradient separation, can sometimes be useful and readily utilized to increase the resolution, although its effect is very modest. The slower flow rate will also decrease the column back pressure. The disadvantage is that when flow rate is decreased, to increase the resolution slightly, there is a corresponding increase in the run time. Method development involves considerable trial and error procedures. Optimization can be started only after a reasonable chromatogram has been obtained. A reasonable chromatogram means that all the compounds are detected by more or less symmetrical peaks on the chromatogram (3,5).
g.) Role of temperature
Temperature variations over the course of a day have quite significant effect on HPLC separations. This can even occur in air conditioned rooms. While temperature is a variable that can affect the selectivity, its effect is relatively small. Also retention time generally decreases with an increase in temperature for neutral compounds but less dramatically for partially ionized analytes. An increase of 1°C will decrease the retention time by 1to 2%. Because of possible temperature fluctuations during method development and validation, it is recommended that the column be thermostated to control the temperature (3,5).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Bioanalytical Method Validation
Bioanalytical method validation (BMV) employed for the quantitative determination of drugs and their metabolites in biological fluids plays a significant role in the evaluation and interpretation of bioavailability, bioequivalence, pharmacokinetic, and toxicokinetic study data. These studies generally support regulatory filings (12). Validation is a tool of quality assurance which provides confirmation of the quality in equipment system, manufacturing processes, software and testing methods. Validation assures that products with pre-determined quality characteristics and attributes can be reproduced consistently within the establish limits (13). Bioanalytical validation is the process of demonstrating that analytical procedures are suitable for intended use. In May 2001, FDA issued a guidance document about validating bioanalytical methodsbased on the deliberations of two workshops: Analytical Methods Validation: Bioavailability, Bioequivalence, and Pharmacokinetic Studies (held on December 3-5, 1990) and Bioanalytical Methods Validation (held on January 12-14, 2000) (14). These guidance take into account a variety of physicochemical, pharmacokinetic, and clinical characteristics for ensuring the validation parameters of bioanalytical methods used for drug and its metabolites analysis (15). Some validation parameters that, at least should be evaluated in a method validation process are : Selectiviy, calibration model, stability, accuracy, and limit of quantification(16).
Need of Validation
Bioanalytical method validation is required to establish, a documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its pre-determined specifications and quality attributes (13). It is essential to employ well-characterized and fully validated bioanalytical methods to yield reliable results that can be satisfactorily interpreted. It is recognized that bioanalytical methods and techniques are constantly undergoing changes and improvements; and in many instances, they are at the cutting edge of the technology. It is also important to emphasize that each bioanalytical technique has its own characteristics, which will vary from analyte to analyte—in these instances, specific validation criteria may need to be developed for each analyte. Moreover, the appropriateness of the technique may also be influenced by the ultimate objective of the study. When sample analysis for a given study is conducted at more than one site, it is necessary to validate the bioanalytical method(s) at each site and provide appropriate validation information for different sites to establish interlaboratory reliability (17).
Benefits of Bioanalytical Method Validation
The validation process provides the highest degree of assurance of the quality of product and process which ought to meet the predetermined quality specification. With this note method validation is also beneficial in many ways (13):
· Decreases the risk of preventing problems and thus assures the smooth running of the process
· Decreases the running cost
· Decreases the risk of regulatory non-compliance
· Fully validated process may require less in-process controls and end product testing (13)
Fundamental Elements of Method Validation
Some elements or points on which any method validation process stands (13)
· Validation plan and Standard operating procedures (SOP)
· Establishment of Acceptance criteria (i.e. testing parameters, limits of acceptability of the components)
· Demonstration of proper equipment system calibration and operation
· Demonstration of meeting of process with an established range of operation for multiple runs
· Demonstration of accuracy and precision for any analytical test methods (13)
Types of Method Validation
It is accepted that during the course of a typical drug development program, a defined bioanalytical method will undergo many modifications. These evolutionary changes (e.g. addition of metabolite, lowering of the lower limit of quantification) require different levels of validation to demonstrate continuity of the validity of the analytical process. Three different types/levels of method validation are defined and characterized as follow (18):
· Full Validation
· Partial Validation
· Cross Validation
a.) Full Validation
· Full validation is important when developing and implementing a bioanalytical method for the first time .
· Full validation is required for a new drug entity
· If metabolites are added to an existing assay for quantification, than full validation of the revised assay will be necessary for the analysis (19).
b.) Partial Validation
Partial Validations are modifications of validated bioanalytical methods that do not necessarily require full revalidations. Partial Validation can range from as little as one intra-assay accuracy and precision determination to a “nearly” Full Validation. Typical bioanalytical method changes which fall into this category include, but are not limited to (19):
· Bioanalytical method transfers between laboratories or analysts.
· Instrument and / or Software Platform changes.
· Change in species within matrix (e.g. rat plasma to mouse plasma).
· Change in matrix within a species (e.g., human plasma to human urine).
· Selectivity demonstration of an analyte in the presence of specific metabolites.
· Selectivity demonstration of an analyte in the presence of concomitant medications.
· Change in analytical methodology (e.g., change in detection systems).
· Change in sample processing procedure(s).
· Rare matrices.
· Change in anti-coagulant in harvesting biological fluid.
· Limited volume changes (e.g. planned pediatric study) (19).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
c.) Cross Validation
Cross Validation is a comparison of two bioanalytical methods. Cross Validations are necessary when two or more bioanalytical methods are used to generate data within the same study. For example, an original validated bioanalytical method serves as the “reference” and the revised bioanalytical method is the “comparator”. The comparisons should be done both ways. Cross validation with spiked matrix and subject samples (18,19):
· Should be conducted at each site or laboratory to establish interlaboratory reliability when sample analyses within a single study is conducted at more than one site, or more than one laboratory.
· Should be considered when data generated using different analytical techniques (e.g. LC-MS-MS vs. ELISA) in different studies are included in a regulatory submission (18,19).
Method Validation Parameters
The fundamental validation parameters are (19,20):
· Selectivity
· Accuracy and Precision
· Calibration curve
· Linearity
· Sensitivity
· Recovery
· Stability
a.) Selectivity
Selectivity should be evaluated to assess the interference at the retention time of the analyte and Internal standard with predetermined method conditions (19,20). Selectivity is the ability of any bioanalytical method to measure and differentiate the analyte in the presence of components, which may be expected to be present (21).
b.) Accuracy and Precision
Accuracy is the closeness of mean test results obtained by the method to the actual value of the analyte. Accuracy is determined by replicate analysis of samples containing known amounts of the analyte (19).
Precision is the closeness of individual measures of an analyte when the procedure is applied repeatedly to multiple sampling of a single homogenous volume of biological matrix (19).
c.) Calibration curve
A calibration curve is the relation between instrument response and know concentrations of the analyte. A calibration curve should be generated for each analyte in the sample. A sufficient number of standards should be used to adequately define the relationship between concentration and response. It is the simplest model used for describing the relation between the responses of instruments and concentrations of analyte (19).
c.) Linearity
The linearity of a bioanalytical method can be defined as the ability to obtain test results directly proportional to the concentration of the analyte in the samples. Linearity is assessed by plotting the measured concentrations against their theoretical values using the data from the intra-day accuracy and precision evaluation (22).
d.) Sensitivity
Sensitivity of a method is defined as the lowest concentration that can be measured with an acceptable limit of accuracy and precision. During this the accuracy and precision at the lowest limit of quantification is determined. These samples should be independent of those used for construction of calibration curve (23). In addition, signal to noise ratio (S/N) will also be calculated to evaluate the noise level. A minimum recommended S/N ration could be 5:1, however acceptance criteria for the signal to noise ratio depend on the individual method (20).
e.) Recovery
Recovery of the analyte from the biological matrix must be determined to ensure adequate and consistent recoveries (24). Recovery is a ratio of the detector response of an analyte from an extracted sample to the detector response of the analyte from an Unextracted sample containing the small amount of the analyte that was added to the extracted sample (23).
g.) Stability
Drug stability in a biological fluid is a function of storage conditions, the chemical properties of the drug, the matrix and the container system (19). Evaluation of stability of targeted analytes in the test biological matrix is critical to validating a bioanalytical method. The integrity of study sample data can be ensured only if supporting stability data are available to confirm that degradation after sample collection has not occurred (25). Different types of stabilities are checked during validation phase :
· Freeze thaw stability
· Short-Term stability
· Long-Term stability
References
1.) Viswanathan CT, Bansal S, Booth B, Destefano AJ, Rose MJ, Sailstad VP, Skelly JP, Swan PG, Weiner R. Quantitative bioanalytical methods validation and Implementation : Best practices for chromatographic and ligand bind assay. The APPS J.2007;9(1):E30-E42.
2.) Shah VP, Midha KK, Findlay JWA, Hill HM, Hulse JD, McGilvery HM, Miller KJ, Patnaik RN, Powell ML, Tonelli A, Viswanathan CT, Yacobi A. Bioanalytical method validation-A revisit with a decade of progress. Pharm. Res.2000;17(12):1551-1557.
3.) Sethi PD. High performance chromatography: quantitative analytical pharmaceutical formulations. 1st edition, 2001. Chapter 1. Introduction. Published by CBS publishers newdelhi; p.3-40.
4.) Singh UK, Pandey S, Pandey P, Keshri PK, Wal P. Bioanalytical method development and validation. Pharm. Exp.2008;2(1):1-8.
5.) Wal P, Kumar B, Bhandari A, Rai AK, Wal A. Bioanalytical method development-Determination of drugs in biological fluids. J Pharm. Sci. Tech.2010;2(10):333-347.
6.) Synder LR, Krikland JJ, Glaich JL. Pratical HPLC method development. 2nd edition,1997. Published by Wiley Interscience Publication, John Wiley & Sons Inc. Canada; p.1-20,100-130.
7.) Zhou S, Song Q, Tang Y, Naidog W. Critical review of development, validation, and transfer for high throughput bioanalytical lc-ms/ms methods. Cur. Pharm. Ana.2005;1:3-14.
8.) Xu X, Lan J, Korftmacher WA. Rapid LC/MS/MS method development for the drug. Ameri. Chem. Soc.2005:A389-A394.
9.) Poole CF. New trends in solid-phase extraction. Trend. Ana. Chem.2003;22(6):362-373.
10.) Yang L, Jensen DS, Vail MA, Dadson A, Linford MR. Direct modification of hydrogen/deuterium-terminated diamond particles with polymers to form reserved and strong cation exchange solid phase extraction sorbents. J Chromato. A.2010;1217:7621-7629.
11.) Sharma GN, Singhal MM, Sharma KK, Sanadya J. Trouble shooting during bioanalytical of drug and metabolites using LC-MS/MS: A review. J Adv. Pharm. Tech. Res.2010;1(1):1-10.
12.) Shah VP. The history of bioanalytical method validation and regulation: Evolution of a guidance on bioanalytical methods validation. The APPS J.2007;9(1):E43-E47.
13.) Rajpoot BS. Validation and process development: A review. Int. Res. J. Pharm.2011;2(1):32-39.
14.) Gao J. Bioanalytical method validation for studies on pharmacokinetics, bioavailability and bioequivalence: Highlights of the FDA’s guideance. Asia. J. Dru. Metab. Pharmaco.2004;4(1):5-13.
15.) Health Canada, Guidance for industry- Conduct and analysis of Bioavailability and Bioequivalence studies – Part A: Oral dosage formulations used for systemic effects, authority of the minister of health, 1992.
16.) Peters FT, Maurer HH. Review: Bioanalytical method validation- How, how much and why. J Chromato.2010;1(1):1-11.
17.) Patil S, Pandurang ND, Kuchekar BS. Bioanalytical method development and validation: Guidelines. Pharm. Info.2009;7(3):1-20.
18.) Tiwari G, Tiwari R. Bioanalytical method validation: An update review. Pharm. Meth.2010;1(1):25-38.
19.) US Food and Drug Administration, Guidance for industry- Bioanalytical methodvalidation, Center for Drug Evaluation and Research, Rockville, MD, 2001. (Available at, http://www.fda.gov/ )
20.) Kalakuntla RR, Kumar KS. Bioanalytical method validation: A quality assurance auditor view point. J Pharm. Sci. Res.2009;1(3):1-10.
21.) Rozet E, Marini RD, Ziemons E, Boulanger B, Hubert P. Advances in validation, risk and uncertainty assessment of bioanalytical methods. J Pharm. Biomed. Ana.2011;55:848-858.
22.) Braggio S, Barnaby RJ, Grossi P, Cugola M. A strategy for validation methods. J Pharm. Biomed. Ana.1996;14:375-388.
23.) Bansal S, Dofestefano A. Key elements of bioanalytical method validation for small molecules. The APPS J.2007;9(1):E109-E114.
24.) Lang JR, Bolton S. A comprehensive method validation strategy for bioanalytical applications in the pharmaceutical industry. J Pharm. Biomed. Ana.1991;9(5):357-361.
25.) Nowatzke W, Woolf E. Best practices during bioanalytical method validation for the characterization of assay reagents and the evaluation of analyte stability in assay standards, quality controls, and study samples. The Apps J.2007;9(2):E117-E122.






