

 ABOUT AUTHOR:
ABOUT AUTHOR:
Kushwaha Anjali *
Naraina vidhya pith, faculty of pharmacy,
Gangaganj, Kanpur
* anjali.kushwaha1@gmail.com
ABSTRACT
Many traditional drugs when given orally exhibit poor and/or highly variable bioavailability and are often accompanied by significant gastro-intestinal side effects. The proprietary powder processing techniques developed by Nanotherapeutics improve the delivery of drugs that cannot normally be taken orally. Nanotherapeutics has developed new inhalation, nasal, injectable, and oral particle delivery systems that:
- Improve the safety and efficacy of low molecular weight drugs,
- Improve the stability and absorption of proteins that normally cannot be taken orally, and
- Extend the life cycle of many existing drug formulations.
An important and exciting aspect in the newly developing field of nanomedicine is the use of nanoparticle drug delivery systems allowing for innovative therapeutic approaches. Due to their small size, these drug delivery systems are promising tools in therapeutic approaches such as selective or targeted drug delivery towards a specific tissue or organ, enhanced drug transport across biological barriers, and intracellular drug delivery. In the my article an overall study on the Nanotherapeutics is done and give a review on the use of Nanotherapeutics in drug delivery methods.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1291
1. Introduction
Colloidal dispersions comprise particles or droplets in the submicron range « l m in an aqueous suspension or emulsion, respectively1. This small size of the inner phase gives such a system unique properties in terms of appearance and application. The particles are too small for sedimentation, they are held in suspension by Brownian motion of the water molecules. They have a large overall surface area and their dispersions provide a high solid content at low viscosity.
Nanoparticles are carriers for conventional drugs as well as for peptides and proteins, enzymes, vaccines, or antigens. According to the process used for the preparation of nanoparticles, nanospheres or nanocapsules can be obtained2. Nanospheres or nanoparticles are homogeneous matrix systems in which the drug is dispersed throughout the particles, whereas nanocapsules are vesicular systems in which the drug is confined to a cavity surrounded by a polymeric membrane.
Nanocarriers for pharmaceutical use can be of polymeric nature or consist of lipophilic components plus surfactants, i.e., liposomes, niosomes, and solid lipid nanoparticles etc.
2. Drug Carrier Systems
|
Generation |
Size |
Definition |
Examples |
|
First |
>1µm |
Abletoreleaseadrugatthetargetsitebut needingaparticulartypeofadministration |
Microspheresandmicrocapsulesfor chemoembolization |
|
Second |
<1µm |
Carriersthatcanbegivenbyageneral routeabletotransportadrugtothetarget site |
Liposomes,nanoparticles,polymerdrug carriers |
|
Third |
<1µm |
Carriersabletorecognizeaspecifictarget |
Monoclonalantibodies;second-generation carrierswithtargetedantibodiesorother ligands |
Table-1: Types of Carriers System
3. Methods of Drug Delivery
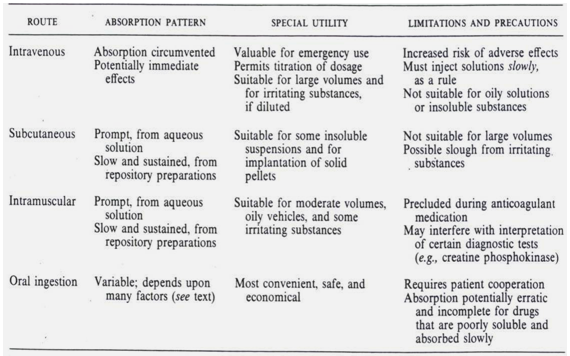
Table-2: Different Types of Delivery Methods
4. Drug Delivery System Technologies
1. Oral Drug Delivery
2. Injection Based Drug Delivery
3. Transdermal Drug Delivery
4. Bone Marrow Infusion
5. Control Release Systems
6. Targeted Drug Delivery:
a. Therapeutical Monoclonal
b. Antibodies
c. Liposomes
d. Microparticles
e. Modified Blood Cells
f. Nanoparticles
7. Implant Drug Delivery System
5. Drug Delivery Carriers
There are different types of drug delivery carriers present which are shown in fig.1.
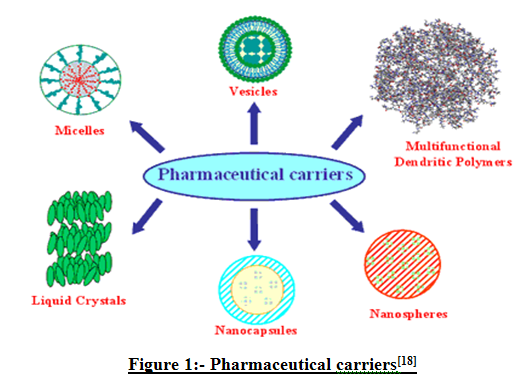
Figure 1- Types of Drug Delivery Carriers
6. Manufacturing of Nanoparticulate Systems
Depending on the nature of the starting material and the intended use of the nanoparticulate system to be prepared, a variety of technologies has been developed and is available for developing and manufacturing colloidal systems3. The following overview provides some background information, the most common methods for drugs to be encapsulated and the most important mechanisms of nanoparticle formation from a physicochemical point of view. Various methods have been developed for preparing nanoparticle dispersions as they are established in industries like coating or plastics. However, the application to pharmaceutical systems containing drugs imposes a number of constraints in selecting the materials, for the size of the particles to be prepared, and for the process itself to prevent e.g. drug degradation. Thus, the methods developed in other disciplines have accordingly been adapted to meet these requirements.
1. Nanosized Drug Substance
Micron-size drug particles are milled in a water-based stabilizer solution for 30-60 minutes to generate nanoparticles with unimodal size distribution5. The amount of the suspension stabilizer is critical since too little of it is unable to prevent aggregation of small particles, and too much of it may accelerate particle growth by Ostwald ripening. Since drug dissolution is directly dependant on the surface area, this approach of increasing the specific surface area might be useful for formulation of drugs with a low solubility in aqueous environments.
Several other methods have been such as the use of supercritical fluid technologies principally leading to particles in the size range of 100 to 500 nm for griseofulvin. Spraying a solution of drug in highly compressed supercritical fluid into atmospheric conditions, rapid expansion of this supercritical solution takes place. Another method to prepare amorphous nanoparticle suspension of poorly water-soluble drugs like Cyclosporine A IS evaporative precipitation into aqueous solution. Rapid evaporation of a heated organic solution of the drug is followed by its atomization into aqueous solution6. This is leads to a nanoparticle suspension, which can be dried to produce oral dosage forms with low crystallinity and small particle
2. Liposomes
Vesicular carriers comprising a hydrophilic core surrounded by one or more lipid bilayer membranes. Liposomes4 can be produced in sizes from below 50 nm up to several /lm depending on the composition and the manufacturing process. They can carry hydrophilic drugs within their core as well as lipophilic substances being dissolved or dispersed in the membrane. A wide variety of manufacturing methods described, of which those using mechanical means to produce the vesicles are preferred for industrial use due to their ability to be well controlled and reproducible. Examples are ultrasound high-pressure homogenization or extrusion through a membrane.
3. Microemulsions
The name "microemulsion" does not refer to them comprising an inner phase in the micrometer range as it might suggest. It is instead most likely derived from their composition being similar to conventional emulsions, albeit they do have distinctly different properties.
Microemulsions comprise two immiscible liquids and at least one emulsifying agent, mostly applied together with a cosurfactant. Systems without an aqueous phase, i.e., surfactant and cosurfactant dissolved or dispersed in oil, are known as self-emulsifying or selfmicro-emulsifying drug delivery systems (SEDDS or SMEDDS). They form microemulsions upon exposure to aqueous media, as in the gastrointestinal tract. Depending on the composition, with an excess of the aqueous phase, no transparent microemulsion is formed, but an opaque conventional emulsion -but without energy input9.
They are stable, recognized as safe by regulatory authorities, and improve drug absorption. For many drugs, they enable the development of alcohol-free formulations. For stabilization, nonionic surfactants are preferred due to their lower toxicity10. However, the use of microemulsions is associated with some drawbacks, limiting their use to the application of problematic drugs rather than being a universal tool.
4. Solid Lipid Nanopartic/es
Colloidal particles consisting of solid triglycerides or other lipid substances were first produced by dispersion of molten lipids by means of high-shear or ultrasound. Similarly, preparation of a microemulsion at higher temperatures can lead to solidification of the lipid phase upon cooling and thus to a dispersion of colloidal lipid particles7, 8.
A method also applicable on larger scale is high pressure homogenization. The process is run either at elevated temperatures with molten lipids in aqueous dispersion or at lower process temperatures where solid lipids are broken down into nanosized particles when pumped through the small gap in the homogenizer.
Solid lipid nanoparticles can alternatively be prepared by rapidly injecting a solution of solid lipids in a water miscible solvent mixture into water to get particles of 80-300 nm. Another group used a particle engineering process of spray-freezing into liquid to generate a rapid dissolving high potency danazol powders of 100 nm Besides the main component, a solid lipid material serving to dissolve or disperse the drug incorporated, SLN often require surfactants for their stabilization, i.e. to prevent aggregation and to enable a nanosized dispersion being generated during processing. Also, these surfactants lead to more round particles, whereas plain lipids generally form cubic crystal-like particle11.
A relatively recent development are the lipid nanocapsules (LNC) prepared by a phase inversion method. This is a solvent-free preparation method leading to small capsules in the size range of 20 to 100 nm.
5. Polymeric Micelles
The capacity of block copolymer micelles to increase the solubility of hydrophobic molecules stems from their unique structural composition, which is characterized by a hydrophobic core sterically stabilized by a hydrophilic corona. Beyond solubilizing hydrophobic drugs, block copolymer micelles can also target their payload to specific tissues through either passive or active means12. Prolonged in vivo circulation times and adequate retention of the drug within the carrier are prerequisites to successful drug targeting. Long circulation times ensue from the steric hindrance awarded by the presence of a hydrophilic shell and the small size (l0100 nm) of polymeric micelles. The self-assembly of amphiphilic block copolymers in water is based on non-polar and hydrophobic interactions between the lipophilic, coreforming polymer chains. Most amphiphilic copolymers employed for drug delivery purposes contain either a polyester or a poly(amino acid)derivative as the hydrophobic segment. Polyethers constitute another class of polymers that can be employed to prepare amphiphilic micelles 13, 14.
Most of the polyether of pharmaceutical interest belong to the poloxamer family, i.e. block-copolymers of polypropylene glycol and polyethylene glycol. Depending on the physicochemical properties of the block copolymer, two main classes of drug-loading procedures can be applied. The first class, direct dissolution, involves dissolving the copolymer along with the drug in an aqueous solvent. This procedure is mostly employed for moderately hydrophobic copolymers, and may require heating of the aqueous solution to bring about micellization via the dehydration of the core-forming segments.
The mechanism by which micelle formation is induced depends on the solvent-removal procedure. For water-miscible organic solvents, the copolymer mixture can be dialyzed against water, whereby slow removal of the organic phase triggers micellization.
Alternatively, the solution casting method entails evaporation of the organic phase to yield a polymeric film where polymer-drug interactions are favoured. Rehydration of the film with a heated aqueous solvent produces drug-loaded micelles. Physical entrapment of a hydrophobic drug may be further achieved through an oil-in-water (OIW) emulsion process which involves the use of a non-water-miscible organic solvent (dichloromethane, ethyl acetate). The above-mentioned techniques all require sterilization and freeze-drying steps to produce injectable formulations with an adequate shelf-life. Process parameters such as the nature and proportion of the organic phase, as well as the latter's affinity for the core-forming segment, can affect the preparation of drug-loaded polymeric micelles and alter the properties of the end product. In addition, the incorporation method itself can modulate the attributes of the yielded micelles15, 16.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
7. Factors Affecting Certain Carrier Properties
To achieve the desired or required properties for nanoparticles to be prepared, an understanding of some general principles of manufacture and composition is beneficial. This allows focused formulation of colloidal drug preparations. The following section provides an overview over the existing data in the field.
a. Drug Loading
Among the influencing factors on the extent of drug loading are method of preparation, additives (e.g. stabilizers, bioadhesives including mucoadhesives, solvent), nature of drug and polymer, their respective solubilites, and pH. Formulation variables can be modulated to increase the drug loading in nanoparticles Depending on both the preparation process and the physicochemical properties of both the drug molecule and the carrier, the drug entrapment can be either by inclusion within the carrier and/or by surface adsorption onto this carrier.
whereas porous nanoparticles may entrap the drug molecule by adsorption either onto the surface or within the macromolecular network. Entrapment within the core of nanocapsules implies the solubility of the drug molecule in the oily phase used during preparation. It should be mentioned that the drug to polymer ratio can be as large as 500: 1 in nanocapsules when this ratio is usually under 10% in nanospheres. Electrical charges on both, the drug molecule and the carrier may influence the loading capacity.
b. Drug Release
Drug release from colloidal carriers is dependent on both the type of carrier and the preparation method applied. As the specific surface area of a nanodispersion is very large, the release rate may be more rapid than from larger structures such as microcapsules. Thus, colloidal carriers are usually not able to act as long-term sustained-release delivery systems.
Depending on the polymer selected and the method of manufacture, drug release from nanoparticles usually follows the following mechanisms: desorption from surface, diffusion through matrix or wall, or erosion of the matrix. In nearly all cases a combination of these phenomena occurs.
Release from nanoparticles may be different according to the drugentrapment mechanism involved. When the drug is adsorbed on the particle surface, the release mechanism can be described as a partitioning process. When the drug is entrapped within the matrix, diffusion plus bioerosion are involved in the release mechanism, whereas diffusion will be the main mechanism if the carrier is not biodegradable.
The active drug can also be bound chemically to a suitable carrier polymer. In such instances, drug release is governed by the cleavage of these chemical bonds, e.g. by hydrolysis or by enzymatically catalyzed reactions. Similar to pro-drug concept, the active moiety is generated after application of the medication.
Special release mechanisms can be procured by selecting polymers having distinct properties with regard to chemical composition and molecular structure. Proper selection of these features like thermo sensitivity or pH-dependency allow to tailor drug release to respond to environmental effects.
c. Stability and Storage
A pharmaceutical formulation faces various stability challenges during preparation, storage and even after administration, before the drug included can be delivered to the targeted site of action. Depending on its chemistry and morphology, a polymer will absorb some water on storage in a humid atmosphere. Absorbed moisture can initiate degradation and a change in physicochemical properties, which can in turn affect the performance in vivo.
Storage conditions may thus be critical to the shelf life of a polymeric nanoparticle formulation. The presence of oligomers, residual monomer, or remaining polymerization catalysts or solvents may impair the storage stability, catalyzing moisture absorption or degradation. The incorporation of drug may also affect the storage stability of a polymer matrix. The relative strength of water polymer bonds and the degree of crystallization of polymer matrix are other important factors. To maintain absolute physicochemical integrity of degradable polymeric drug delivery device, storage in an inert atmosphere is recommended17.
Freeze-drying is a good method to dry nanoparticles in order to increase the stability of these colloidal systems. However, due to their vesicular nature, especially nanocapsules are not easily lyophilized, as they tend to collapse, releasing the core content.
d. Nanoparticle-containing Dosage Forms
For oral delivery into the human body nanoparticles can be also administered as their aqueous dispersion as the final dosage form. This is a way of delivery without further processing after nanoparticle formation. However, poor stability of the drug or polymer in an aqueous environment or poor taste of the drug may require the incorporation of the colloidal particles into solid dosage forms, i.e. into capsules and tablets18.
Colloidal particles can be incorporated into solid dosage forms either in solid or liquid form. The dispersion of the colloidal particles can be dried if needed together with suitable excipients, followed by filling of the dried powder into capsules or compressing it into tablets. Suitable conventional excipients such as fillers or binders can be added to adapt the processability of the dried nanoparticle dispersion or to tailor the final dosage form.
Alternatively, the aqueous dispersion of the colloidal particles can be incorporated into the solid dosage form as a liquid, for example by granulation of suitable fillers with the colloidal dispersion to form a granulation. Such granules can subsequently be filled into capsules or be compressed into tablets19, 20. The mechanical stress applied during drying and/or compression of nanoparticle-based formulations has to be considered when selecting a process for transforming nanoparticle dispersion into a solid dosage form.
8. Application of Nan particles
· Oral Applications
· Inhaled / Nasal Applications
· Topical Applications
· Injectable Applications
Oral Applications
Typically, tablets or capsules are used to deliver greater than 90% of pharmaceuticals to patients. Factors that affect the efficacy of a drug include the solubility, bioavailability, biological half-life, dose and dosing regimen, and shelf-life, as well patient factors including diet and health condition. Technologies that can improve oral delivery of drugs by controlling the release and absorption in the GI tract are in great demand, and improved powder processing is one strategy that can overcome these obstacles. In vivo studies have shown that sustained-release and improved bioavailability of pressed powders for improved tablet processing of next generation pharmaceuticals.
Nanotherapeutics particle delivery systems are able to:
· Improve the oral bioavailability of insoluble and poorly absorbed drugs that require injection (antibiotics, antivirals, anti-inflammatories, and nutritionals).
· Enable oral delivery of macromolecules (peptide/protein).
· Provide pharmaceutical life-cycle management opportunities.
Inhaled / Nasal Applications
Novel dry powder formulations for inhaled or nasal delivery have been produced. They have exhibited improved therapeutic endpoints compared to conventional dry or liquid formulations for low molecular-weight compounds for local or systemic delivery; biologically active peptides; and vaccines against infectious pathogens. Formulation and delivery of novel nanoparticles offers advantages, including:
· higher shelf-life stability
· better control of particle size and deposition efficiency
· control of cell uptake and targeting
· controlled release-rates
· increased systemic bioavailability
Topical Applications
Nanoparticles in topical gels and creams are being developed as an alternative carrier system to emulsions and liposomes. Solid lipid nanoparticles (SLN), produced by exchanging a portion of the liquid oil part of an emulsion with a lipid solid at room temperature, have been shown to deliver molecules deeper into the skin than traditional mixtures. Formulation and delivery of novel nanoparticles and microparticles for topical delivery offers several advantages including:
* Increase in loading capacity.
* Potential for decreased skin irritation.
* Stabilization of sensitive actives.
* Controlled-release.
* Physical and chemical long-term stability.
Injectable Applications
Nanoparticle and microparticle suspensions offer an advanced approach to the delivery of insoluble drugs and formulations for sustained release. Advantages include:
· Low excipient loads
· Smaller dose volumes
· Better control of particle size and dispersion efficiency
· Controlled release-rates
· Capability for sterile filtering
· Increased efficacy and systemic bioavailability
9. Requirements of Nano therapeutic Applications
· Devices should be non-invasive
· Devices target therapeutics payloads to site of disease.
· Devices should maximize therapeutic benefit and minimize undesired side effect
10. Purpose of Nano-therapeutics Discussions
· Focused on nanodevices rather than nanomaterials.
· Purpose- serves as integral component of drug delivery or other clinical devices.
· Incorporating biological structure into nanobiological devices: special challenge with traditional engineering design etc.
11. Conclusion
Nanotherapeutics are of particular importance since there will be hundreds of new bio-engineered drugs entering the marketplace in the next few years that cannot be absorbed orally using traditional systems because of enzymatic breakdown. Sustained-release delivery systems have shown considerable promise within the industry since most peptides/proteins have short systemic half-lives and require that they be given by frequent injection or constant infusion, a situation generally not preferred by either patients or physicians. Thus the use of Nanotherapeutics in new drug delivery system is increases day by day and also gives best results.
12. References
1. Abdelwahed, W., Degobert, G. and Fessi, H. (2006). Investigation of Nanocapsules Stabilization by Amorphous Excipients during Freeze-drying and Storage. Eur. Pharm. Biopharm., 63, pp. 87-94.
2. Allemann, E., Gumy, R. and Doelker, E. (1993 a). Drug-loaded Nanoparticles Preparation Methods and Drug Targeting Issues. Eur.Pharm. Biopharm., 39, pp.I73-191.
3. Bangham, A. D., Standish, M. M. and Watkins, J. C. (1965). Diffusion of Univalent Ions across the Lamellae of Swollen Phospholipids. Mol. Biol., 13, pp. 238-252.
4. Barenholz, Y., Amselem, S. and Lichtenberg, D. (1979). A New Method for Preparation of Phospholipid Vesicles (Iiposomes) -french press. FEBS Lett. 99, pp. 210-214.
5. Bodmeier, R. and Chen, H. (1990). Indomethacin Polymeric Nanosuspensions Prepared by Microfluidization. J. Control. ReI., 12, pp. 223-233.
6. Bodmeier, R. and Paeratakul, O. (1989). A Novel Approach to the Oral Delivery of Micro-or Nanoparticles. Pharm. Res., 5, pp. 413-417.
7. Chen, H. and Langer, R. (1998). Oral Particulate Delivery: Status and Future Trends. Adv. Drug Del. Rev., 34, pp. 339-350.
8. Chen, X., Young, T. J., Sarkari, M., Williams, R. 0. Johnston, K. P. (2002). Preparation of Cyclosporine A Nanoparticles by Evaporation Precipitation into Aqueous Solution. Int. J. Pharm., 242, pp. 3-14.
9. Constantinides, P. P. (1995). Lipid Microemulsions for Improving Drug Dissolution and Oral Absorption: Physical and Biopharmaceutical Aspects. Pharm. Res., 12, pp. 1561-1572.
10. Couvreur, P., Kante, B., Roland, M., Guiot, P., Bauduin, P. and Speiser, P. (1979). Polycyanoacrylate Nanocapsules as Potential Lysosomotropic Carriers: Preparation, Morphological and Sorptive Properties. J. Pharm. Pharmacal., 31, pp.331-332.
11. De Chasteigner, S., Cave, G., Fessi, H., Devissaguet, J.P. and Puisieux, F. (1996). Freeze-Drying of Itraconazole-Loaded Nanosphere Suspension: A Feasibility Study. Drug Dev. Res., 38, pp. 116-124.
12. De Jaeghere, F., Allemann, E., Feijen, J., Doelker , E. and Gumy, R. (1999). FreezeDrying of PLA-PEO Nanoparticles -from Basic Principles to Rational Optimization. Proceed. Int. Symp. Control. ReI. Bioact. Mater. 26, pp. 709-710.
13. De Kruijff, B., Cullis, P. R., Radda, G. K. (1975). Outside-inside Distributions and Sizes of Mixed Phosphatidyl-Cholesterol Vesicles. Biochim. Biophys. Acta, 436, pp.729-740.
14. Eerikainen, H., Watanabe, W., Kauppinen E. and Ahonen, P. P. (2003). Aerosol Flow Reactor Method for Synthesis of Drug Nanoparticles. Eur. 1. Pharm. Biopharm., 55, pp. 357-360.
15. Fessi, H., Puisieux, F., Devissaguet, J. P., Ammoury, N. and Benita, S. (1989). Nanocapsule Formation by Interfacial Polymer Deposition Following Solvent Displacement. Int. 1. Pharm., 55, pp. RI-R4.
16. Freitas, C. and MUller, R. H. (1998). Effect of Light and Temperature on Zeta Potential and Physical Stability in Solid Lipid Nanoparticle (SLNTM) Dispersions. Int. 1. Pharm., 168, pp. 221-229.
17. Gupta, R. B. (2006 b). Supercritical Fluid Technology for Particle Engineering, in Gupta, R.B., Kompella, U.B. (eds.), Nanoparticle Technology for Drug Delivery, , pp. 53-84.
18. Lehmann, K. O. R. (1997). Chemistry and Application Properties of Polymethacrylate Coating Systems, in: McGinity, J.W. (ed.), Aqueous Polymeric Coatings for Pharmaceutical Applications (Marcel Dekker Inc., New York, Basel, Hong Kong), pp. 101-176.
19. Mayer, L. D., Hope, M. J. and Cullis, P. R. (1986). Vesicles of Variable Sizes Produced by a Rapid Extrusion Procedure. Biochim. Biophys. Acta, 858, pp. 161-168.
20. Schroeder, U., Sommerfeld, P, Ulrich, S. and Sabel, B. A. (1998). Nanoparticle Technology for Delivery of Drugs across the Blood-Brain Barrier. I. Pharm. Sci., 87, pp. 1305-1307.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






