

About Author: Vikas Mahajan* (M.Pharm 1st year), Naiyer Shahzad, Sachin Mager
Singhania University,
Pacheri Bari, Rajasthan - 333515
Reference ID: PHARMATUTOR-ART-1054
Abstract
In this study Cytotoxic potential of two Indian medicinal plant extracts were investigated. The invitro cytotoxic potentiality investigated as the ability of these two extracts to inhibit tumour cell line growth with help of MTT & Trypan blue assay. With this investigation we had also focused on angiogenesis. The cell lines studied are MCF7, A549 and DU145 with the methanolic extract of Alium sativum (MEAS) (garlic) and Emblica officinalis (MEEO) (amla). Both drugs are extracted by maceration method. The extract were concentrated & dissolved in DMSO Solvent & stock solution is prepared of conc. 1mg/10 ml. from which different conc. are prepared as 100, 10, 1, 0.1,0 .001 µg/ml. Both plant extracts preapared concentration are exposed in MCF7, A549 & DU145 in 96 well plate in which MTT dye was added later & allow it for 96 hr. after incubation period absorbance was taken in spectrophotometer at 517nm. With same trypan blue also performed & Counting of cell was done on inverted microscope. Results indicates that the above plant extracts hsowing anticancer and antiangiogenesis activity of Same plant extract were studied on tube formation cell based models also. From this study we can conclude that both drugs possess anticancer activity for MCF7, A549 & DU145 for different concentrations.
[adsense:336x280:8701650588]
Introduction
Cancer is a disease in which a group of abnormal cells grow uncontrollably by disregarding the normal rules of cell division. Normal cells are constantly subject to signals that dictate whether the cell should divide, differentiate into another cell or die. Cancer cells develop a degree of autonomy from these signals, resulting in uncontrolled growth and proliferation. If they proliferate and allowed to continue and spread, it can be fatal. In fact, almost 90% of cancer-related deaths are due to tumour spreading – a process called metastasis. All mammalian cells share similar molecular networks that control cell proliferation, differentiation and cell death. The prevailing theory, which underpins research into the genesis and treatment of cancer, is that normal cells are transformed into cancers as a result of changes in these networks at the molecular, biochemical and cellular level, and for each cell there is a finite number of ways this disruption can occur. Phenomenal advances in cancer research have given us an insight into how cancer cells develop this autonomy. These changes (DNA mutations) produce proteins that disrupt the delicate cellular balance between cell division and quiescence, resulting in cells that keep dividing to form cancers[1]. Some people inherit damaged DNA, but in most cases people damage it themselves through lifestyle choices such as smoking, exposure to ultraviolet radiation (UV) from the sun or exposure to cancer-causing substances known as carcinogens in the environment, such as asbestos [2]. While being infected with certain viruses, such as the human papillomavirus (HPV) and human immunodeficiency virus (HIV), can increase the risk of some types of cancer cancer is not contagious. You can't "catch" cancer from someone who has the disease. Scientists also know that an injury or bruise does not cause cancer [3,4]. Prophylactic vaccines have been developed to prevent infection by oncogenic infectious agents such as viruses, and therapeutic vaccines are in development to stimulate an immune response against cancer-specific epitopes [5].
2. Angiogenesis:
Only a few years ago, it was proposed that solid malignant tumours should be primary targets for antiangiogenic treatment; in fact, three options have been considered so far:
1. Inhibition of the turnover from an avascular primary tumour into a fully vascularized tumour (quite frequently, this will not be feasible because avascular primary tumours have a diameter of only a few millimetres and will usually escape clinical detection)
2. Slowdown of tumour progression by preventing a tumour from becoming highly vascularized
3. Prevention of neovascularization of distant metastases.
The process of forming new blood vessels is known as angiogenesis. Blood vessel formation is of two types. Vasculogenesis is the generation of blood vessels from endothelial cell progenitors (hemangioblasts). It is responsible for the formation of the primary vasculature of the body during early embryonic development [6]. Angiogenesis is a complex, highly regulated process, involving the sprouting, splitting, and remodeling of the existing vessels. Physiologically angiogenesis occurs under tight regulation in the female reproductive system and during wound healing. Many pathological conditions such as ischemic tissue injury are also benefited by revascularization. On the other spectrum, excessive angiogenesis, may result in different diseases including cancer, atherosclerosis, rheumatoid arthritis, Crohn's disease, diabetes, psoriasis, endometriosis, and adiposity [7]. These diseases may benefit from the therapeutic inhibition of angiogenesis[7].
3. Plant-derived tumour inhibitors:
Plant materials have been used in the treatment of malignant disease for centuries; a comprehensive survey of literature describing plants used against cancer listed over 1400 genera. (Hartwell, 1967) The following summary has been compiled from Fundamentals of Pharmacognosy and phytotherapy by Michael Heinrich 2004. Recent phytochemical examination of plants which have a suitable history of use in folklore for the treatment of cancer has indeed often resulted in isolation of constituents with antitumour activity. From the time of Galen (about AD 180), the juice expressed from woody nightshade (Solanum dulcamara) has been used to treat cancers, tumours and warts, and references to its use have appeared in the literature of many countries. The active tumour inhibitory principle has been identified as the steroidal alkaloid glycoside, beta solamarine. Similarly, many centuries ago, the druids claimed that mistletoe (Viscum album) could be used to cure cancer; protein fractions with marked antitumour activity have been isolated from mistletoe extract. Mezereon (Daphne mezereum), despite its toxic properties, has also been used in many countries for the treatment of cancer. The active antitumour constituent of this plant has been identified as a diterpene derivative mezerein, which is structurally very similar to the toxic principle daphnetoxin. Various lichens, e.g. species of Cladonia, Cetraria and Usnea, also have a history of use in folk medicine against cancer since about AD970. These are all rich sources of usnic acid, a compound which has been recognized for many years as an antibacterial and antifungal agent but only more recently as an antitumour compound. The most successful of the higher plants used in cancer chemotherapy are the alkaloids of Catharanthus roseus. Research on this plant was stimulated by its mention in folklore, not as a cure for cancer but in the treatment of diabetes. No hypoglycaemic activity was detected but treated test animals became susceptible to bacterial infections, and this led the researchers to undertake extensive examination for possible immunosuppressive principles for this effect. A number of bisindole alkaloids showing antileukaemic activity has been subsequently isolated and two of these, vincristine and vinblastine, are now extracted commercially from Catharanthus roseus.
4. Material And Methods [8-10]
4.1 Reagents
Alcohol 70%, MEM media (Minimal essential media), Trypsin, MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, a tetrazole)., Trypan blue, Distilled water, etc.
4.2 Instrument
Autoclave, N2 liquid container, CO2 incubator, Inverted microscope. Stereosome microscope, Filtration assembly, Hemocytometer, Centrifuge, Micropippete, etc.
4.3 Extraction method
A wide mouth bottle or other container which can be well stoppered is used for maceration process. Closed container is essential to prevent the evaporation of menstruum or solvent which is mostly concentrated alcohol.
[adsense:336x280:8701650588]
4.3.1 Method
• Weigh 20 gm of drug and transfer it into container containing menstruum and cover it with help of cotton or other closure which is suitable one.
• Keep the it for adequate duration of time.
• Shaking of drug during maceration is essential inorder to replace the saturated layers around the drug with fresh menstruum.
• After straining, the marc is pressed in filter press, hydraulic press or hand press.
• Then go for concentration of drug.
5. Cell Culture And Cell Line:
Cell culture is a techniques by which prokaryotic or eukaryotic cells are grown under controlled condition. Isolation of cells from tissue for exvivo culture in different maner, Mononuclear cells can remove by digestive enzyme like trypsin,pronase tissue transfer in media it termed as explant culture.
5.1 Main steps in Cell Subculture
Step1) After subculture, cells are sparsely attached to the cell culture flask/plate.
Step 2) After a few hours-days (depending on the cell line), cells grow into the gaps of the plate, and reach confluency. At this stage, they need to be split.
Step 3) Addition of trypsin to the plate after washing away the serum digests away the protein-cell contacts allowing the cells to break away from their contacts.
Step 4) After trypsinization, most of the cells are removed from contact with the plate, and are floating.
Step 5) After subculture, most of the cells are removed and either discarded, or added to other flasks. Fresh Fetal bovine serum (FBS) and media is added
5.2 Selection and isolation of Cell line [11]:
? Growth characteristics
? Species
? Finite Vs continuous
? Control cell line
? Validation
Human cancer cells are isolated from the patients and characterized at cellular and molecular levels. Isolated cells are cultivated in specialized mediums and specialized incubators to provide them physiological conditions required for the growth.
5.3 Cell Line:
The subculturing of the primary culture gives rice to cell lines. The term continuous cell line implies the indefinite growth of the cell in the subsequent subculturing. On the other hand, finite cell lines represent the death of cell after several subcultures [12] . So the seleated cell lines are DU145, A549, MCF7 name as prostate cancer cell line, lung cnacer and breast cancer respectively.
5.3.1 cell counting protocols:
Traditionally a cell culture is counted prior to plating the cells or for culturing the cells in flasks, roller bottles, etc. This is to determine the cell number of viable cells. Counting cells consistently is important to the responsiveness of the cells. Counting cells can be done numerous ways. A common way of counting cells is by using a hemacytometer and light microscope. This method can be subjective and is time consuming. There are also several automated methods of counting cells including the Coulter Counter or the Vi-CELL. The basis of the Coulter Counter is detects changes in electrical conductance of a small aperture as fluid containing the cells flows past the detector. The deflection is then detected as a particle or cell. The Vi-CELL is an automated method of counting cells using the trypan blue cell exclusion method.
5.3.2 Routine Maintenance [13]:
? Subculture
? Seeding
? feeding
? Cryopreservation
5.3.3 Protocols [14]:
5.3.3.1 Subculture
• Prepare laminar air flow (LAF).
• Flame burner first and give flame to T-25 flask.
• Open T-25 flask and give flame to it and needle.
• Remove spent media with help of liquid waste handling system (LWHS).
• Then take adequate amount of trypsin wash it and remove it with help of LWHS.
• Then again add trypsin for detaching cells and keep it for incubation for appropriate duration as per confluency of flask.
• Then add adequate amount media into it and transfer half amount of media in another T-25 flask.
• Transfer both flask to co2 incubator.
5.3.3.2 Feeding
• Prepare laminar air flow.
• Flame burner first and give flame to T-25 flask.
• Open t-25 flask and give flame to it and needle.
• Remove media from flask with help of liquid waste handling system (LWHS).
• Add same amount of media in flask and transfer it to co2 incubator.
5.3.3.3 Seeding
• Prepare laminar air flow.
• Flame burner first and give flame to T-25 flask.
• Open T-25 flask and give flame to it and needle.
• Remove media from flask with help of liquid waste handling system (LWHS).
• Then take adequate amount of trypsin wash it and remove it with help of LWHS.
• Then again add trypsin for detaching cells and keep it for incubation for appropriate duration as per confluency of flask.
• Then add adequate amount media into it and make suspension and transfer it into petridish.
• Add it in 96 well plate as per.
5.3.3.4 Cryopreservation
• It is a process where cells or whole tissues are preserved by cooling to low sub-zero temperatures, such as (typically) 77 K or −196 °C (the boiling point of liquid nitrogen).
• At these low temperatures, any biological activity, including the biochemical reactions that would lead to cell death, is effectively stopped. However, when cryoprotectant solutions are not used, the cells being preserved are often damaged due to freezing during the approach to low temperatures or warming to room temperature Pipette, 50 ml. tube, Burner, Cryo vial.
• Laminar arrangement (switch on the U.V. for 20-30 min.). Water bath set at 37 ºC. media taken out from the freezer for melting. Wiped the media tube & kept inside the laminar. 2 ml. media was left in the flask and sucked out rest of the media & poured into the 50 ml. tube.
• Centrifuged the cells at 1000 rpm for 10 min. Then removed the suspended media through liquid waste handling system.
• Then 2ml. fresh media was added in the 50 ml. tube mixed well & transferred to the cryo tube. 25μl DMSO for preservation were added.
• Then it was kept inside the fumes of the liquid nitrogen container. Then added 3 ml. media in the flask & the flask was transferred inside the CO2 incubator.
5.3.3.5 Cryorevival
• Pipette, Petri dish, 50 ml tube, Burner.
• Filled 5ml. water in a 50 ml. tube for centrifugation. Switched on the water bath for & set the temp. at 37 ºC.
• The cryo vial was taken out from the liquid nitrogen container. The vial in the water for melting (but not completely melted) was kept.
• Transferred the cell suspension in the 50 ml. tube from the vial. Vol. up to 5ml was done. Then centrifuged the suspension for 10 min. at 1000 rpm. Cell suspension was transferred into Petri dish. It was kept inside the incubator.
6. Assay Performed:
6.1 MTT Assay
6.1.1 Introduction [15]
MTT assay is based on conversion of the MTT dye to a purple coloured formazan crystal by the active mictochondrial reductases present in the viable cells. The purple colour thus formed is directly proportional to the viable cells present. This provide study of the cytotoxic activity of the test compounds.
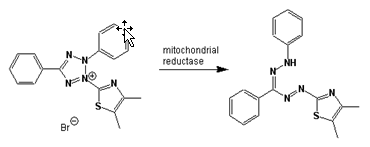
Fig.6.1a Yellow MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, a tetrazole) is reduced to purple formazan in living cells.
6.1.2 Principle
The MTT assay is laboratory tests and standard colorimetric assays (an assay which measures changes in color) for measuring the activity of enzymes. It can also be used to determine cytotoxicity of potential medicinal agents and other toxic materials, since those agents would result in cell toxicity and therefore metabolic dysfunction and therefore decreased performance in the assay. Yellow MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, a tetrazole) is reduced to purple formazan in living cells.A solubilization solution (usually either dimethyl sulfoxide, an acidified ethanol solution, or a solution of the detergent sodium dodecyl sulfate in diluted hydrochloric acid) is added to dissolve the insoluble purple formazan product into a colored solution. The absorbance of this colored solution can be quantified by measuring at a certain wavelength (usually between 500 and 600 nm) by a spectrophotometer. The absorption maximum is dependent on the solvent employed.
6.1.3 Method
Make four groups
1. + control = standard drug (Paclitaxel/Doxorubicin)
2. – control = Test drug + media
3. + Test = Test drug + Media + Cell line
4. Blank = Media or Blank
1. Make dilution of concentration 100 µg/ml, 10 µg/ml, 1 µg/ml, 0.1 µg/ml, 0.01 µg/ml from stock solution(test drug+DMSO) having concentration 10mg/ml.
2. Make normal count on haemocytometer before seeding the cells in plate
3. Add 10µl from each conc. in 4wells i.e. 20 wells for one drug.
4. Plate should contain 5,000-10,000 cells/ml into each well of 96-well culture plate.
5. Incubate the cells for 96 hr in CO2 incubator.
6. After it cells are incubated with basal medium containing 0.5 mg/ml MTT in CO2 incubator at 37 ?C for appropriate duration of time.
7. The medium is aspirated, and the formazan product is solubilized with dimethyl sulfoxide (DMSO).
8. Absorbance at 570 nm is measured for each well using a microplate reader on colorimeter.
9. Analyse data of test with standard drug and plot graph.
6.2 Trypan blue assay
6.2.1 Introduction [16,17]
Trypan blue is derived from toluidine, that is, any of several isomeric bases, C14H16N2, derived from toluene. Trypan blue is so-called because it can kill trypanosomes, the parasites that cause sleeping sickness. An analog of trypan blue, suramin is used pharmacologically against trypanosomiasis. Trypan blue is also known as diamine blue and Niagara blue.Trypan blue is commonly used in microscopy (for cell counting) and in laboratory mice for assessment of tissue viability. The method cannot distinguish between necrotic and apoptotic cells
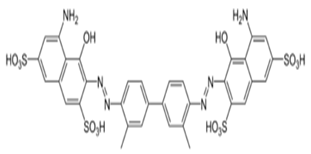
Fig. 6.2a Trypan blue
6.2.2 Principle
Trypan blue is vital stain used to selectively colour dead tissues or cell blue. It is diazo dye. Live cells or tissues with intact cell membrane are not colourred. Since cells are very selective in compounds that pass through membrane, in viable cell trypan blue is not absorbed, however it transverse the membrane in dead cell. Hence dead cells are shown distinct blue colour in microscope. Since live cells excluded from staining, hence this staining method is also called as dye exclusion method.
6.2.3 Method [18]
1. Take cell suspension being tested on petridish mix with 0.4% trypan blue.
2. Allow mixture to incubate 3 min at room temperature.
3. Cells should be counted within 3 to 5 min of mixing with trypan blue, as longer incubation periods will lead to cell death and reduced viability counts.
4. Mixing can be performed in a well of a microtiter plate or a small plastic tube using 10 to 20 μl each of cell suspension and trypan blue.
5 Apply a drop of the trypan blue/cell mixture to a hemocytometer . Place
the hemocytometer on the stage of a binocular microscope and focus on the cells.
6. Count the unstained (viable) and stained (nonviable) cells separately in the hemocytometer.
7. Calculate the percentage of viable cells by using following formula-
viable cells (%) = total number of viable cells per ml/ total number of cells per ml *100
7. Results And Disscusion
Table 7.1a Results of MTT and Trypan Blue assay
|
Cell line |
MCF7 |
A549 |
DU145 |
|||
|
Sample code |
A |
B |
A |
B |
A |
B |
|
IC50 (µg/ml) |
06 |
30 |
20 |
05 |
02 |
30 |
|
Trypan Blue |
44 |
53 |
44 |
53 |
48 |
46 |
(A)=Garlic,(B)=Amla
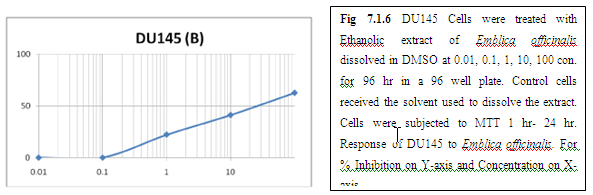
(A) GARLIC
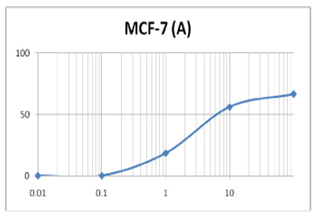
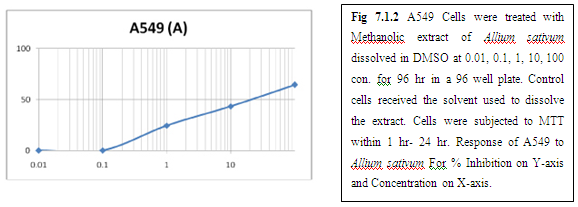
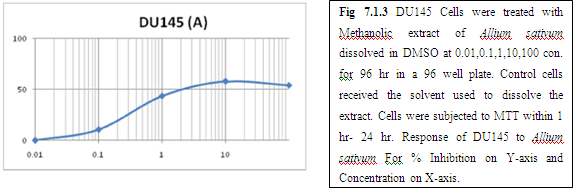
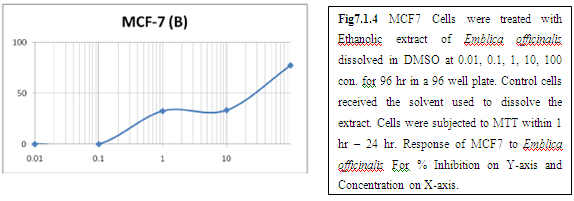
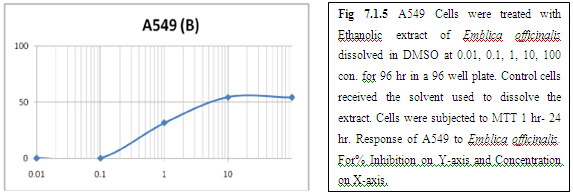
(B) AMLA
Trypan Blue Exclusion Assay Result
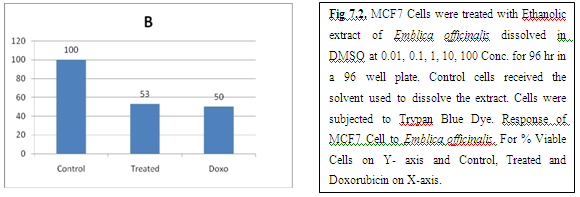
(A) GARLIC
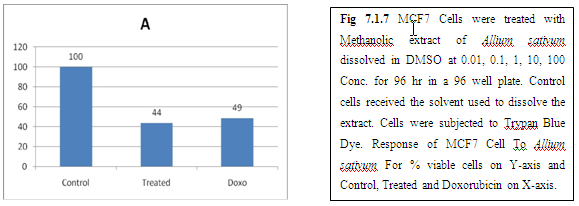
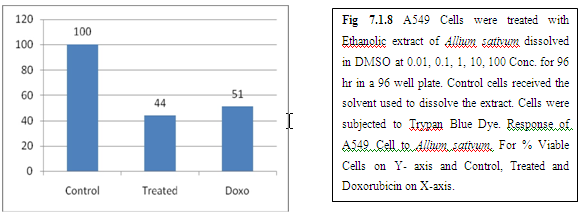
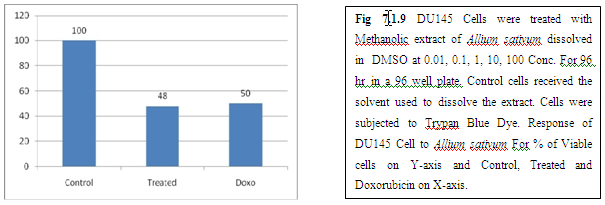
(B) AMLA
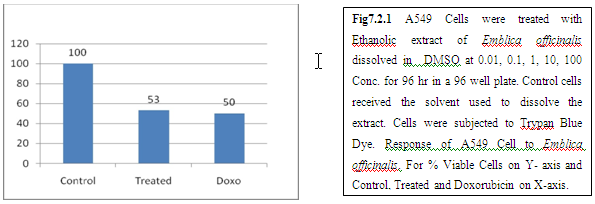
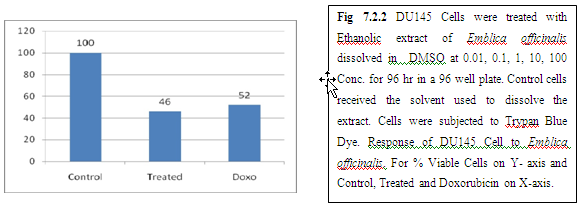
9. Discussion And Conclusion:
From above graph IC50 value of garlic for MCF7, A549, DU145 is less as compare to amla for MCF7, A549, DU145, from this it may concluded that Emblica officinalis (Amla) shows potent cytotoxicity than Allium sativum (garlic) for above cell line.
And in case of Trypan blue the cytotoxic acton of Allium sativum showing more potent activity than Emblica officinalis as treated drug shows more inhibition of cells than standard doxorubicin for same cell line.
Invitro cytotoxicity tests currently use in applied in developmental stages of anticancer drug discovery are able to select the most potent compounds. Angiogenesis plays an important role in the growth and spread of cancer. New blood vessels “feed” the cancer cells with oxygen and nutrients, allowing these cells to grow, invade nearby tissue, spread to other parts of the body, and form new colonies of cancer cells.Above graphs and figures shows the how angiogenesis occur and inhibition of vascularisation. Research in angiogenesis offers a potential to cure a variety of diseases such as Alzheimer's and AIDS.
The graphs and according to above results and discussion it may concluded that the Allium sativum (garlic) having more cytotoxicity than Emblica officinalis (Amla) for MCF7, DU145 and A549 cell lines.
References:
1. Momna Hejmadi and Ventus introduction to cancer biology (2009) ApS ISBN 978-87-7681-478-6.
2. Caius J. F. Medicinal and Poisonous Plants of India, Scientific Publishers, Jodhpur (1986)
3. Singh B. N; Sharma, P.V; "Effect of cance"r. J. Res. Ind. Med., 1971; 5 : 223
4. Baruchin AM, Sagi A, Yoffe B, Ronen M. study on cancer. 2001 Nov;27(7):781
5. "Cancer Vaccine Fact Sheet". NCI. 2006-06-08. Retrieved 2008-11-15.
6. Hayden, Erika C. (2009-04-08). "Cutting off cancer's supply lines". Nature 458 (7239): 686–687. doi:10.1038/458686b. PMID 19360048.
7. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995; 1:27-31.
8. Freshney R, Culture of animal cells manual of basic technique, 4th edition (2000) 181-184.
9. www.promega.com/paguid/chap4.htm title4
10. Mosmann T (December 1983). "Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays" .Journal of immunological methods 65 (1-2): 55–63. PMID 6606682.
11. Cory AH, Owen TC, Barltrop JA, Cory JG (July 1991). "Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture". Cancer communications 3 (7): 207–12. PMID 1867954.
12. Boyd MR, Paull KD. Some practical considerations and applications of the National Cancer Institute In vitro anticancer drug discovery screen. Drug Dev Res 1995;34:91 – 109.
13. Holbeck SL. Update on NCI in vitro drug screen utilities. Eur J Cancer 2004;40:785– 93.
14. Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990;82:1107– 12.
15. Shapiro, H.M. 1988. Practical Flow Cytometry, 2nd ed., p. 129. John Wiley & Sons, New York.
16. Brawley OW (2004). "Prostate cancer screening: clinical applications and challenges". Urol. Oncol. 22 (4): 353–7.
17. Bedard PL, Krzyzanowska MK, Pintilie M, Tannock IF (2007). "Statistical power of negative randomized controlled trials presented at American Society for Clinical Oncology annual meetings". J. Clin. Oncol. 25 (23): 3482–7.






