

About Authors:
Hardwari L. Yadav, P.K. Singour, R.S. Pawar, U. K. Patil
Department of Pharmaceutical Chemistry,
VNS Institute of Pharmacy,
Neelbud, Bhopal (M.P.)
Abstract
Chalcone are the important constituent of many natural sources and have variety of biological activities. The novel chalcone derivatives isolated from the fruits of Mallotus philippinensis called mallotophilippens. The chalcones are synthesize by claisen-schmidt base catalyzed condensation of appropriate aromatic ketones or substituted aromatic ketones with benzaldehyde or substituted benzaldehydes. Chalcones, considered as the precursors of flavonoids and isoflavonoids, are abundant in edible plants, and have also been shown to display a diverse array of pharmacological activities. Recent reports suggest antimicrobial activity of chalcones (antibacterial and antifungal), as well as antileishmanial, antimalarial, antiviral and anti-inflammatory activities of these compounds.
[adsense:336x280:8701650588]
Reference ID: PHARMATUTOR-ART-1164
Introduction
Chalcones considered as the precursors of flavonoids and isoflavonoids are abundant in edible plants and have also been shown to display a diverse array of pharmacological activities. Chalcones or 1, 3-diaryl-2-propen-1-ones belong to the flavonoid family. Chemically they consist of open-chain flavonoids in which the two aromatic rings are joined by a three-carbon α, ß-unsaturated carbonyl system. Chalcones have been reported to possess many useful properties, including anti-inflammatory, antimicrobial, antifungal, antioxidant, cytotoxic, antitumor and anticancer, antimalarial, antileishmanial, antidiabetic activities. The presence of α β unsaturated ketone group in chalcone is responsible for their different activities.1, 2
A number of chalcone derivatives, have also been found to inhibit several important enzymes in cellular systems, including xanthine oxidase, aldose reductase, epoxide hydrolase, protein tyrosine Kinase,and quinone reductase.3
2. Anti-inflammatory properties
Inflammatory stimulation, macrophages produce NO, prostanoids and proinflammatory cytokines such as interleukin (IL)-1b and TNF-α. NO is generated by NO synthase (NOS) and induces tissue injury at the inflammatory site. Three isoforms of NOS have been identified; endothelial (eNOS), neuronal NOS (nNOS) and inducible NOS (iNOS). Among the three, iNOS is expressed in response to various inflammatory stimuli and causes a large amount of NO to be produced by macrophages during the inflammatory process. TNF-α is one of the most important proinflammatory cytokines and is produced mainly by activated monocytes and macrophages. It induces various biological responses including tissue injury, shock and apoptosis. TNF- α also induces the secretion of cytokines such as IL-1, IL-6 and IL-10, and activates T cells and other inflammatory cells.6
[adsense:468x15:2204050025]
The inhibition of prostaglandin E2 (PGE2) and nitric oxide (NO) production has been proposed as a potential therapy for different inflammatory disorders. Large amounts of NO may lead to tissue damage. In inflammatory diseases such as rheumatoid arthritis, excessive NO production by activated macrophages has been observed. Therefore, it would be interesting to develop potent and selective inhibitors of NO for potential therapeutic use. Chalcone derivatives show anti-inflammatory activity by inhibit the cyclooxygenase (Cox), nitric oxide synthase enzyme and tumor necrosis factor-α.1, 4, 5
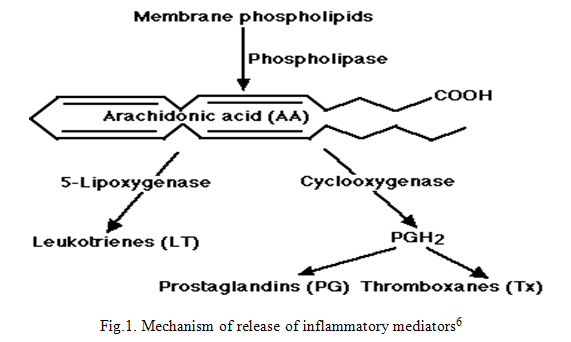
The pathway of release of different chemical mediators responsible for inflammation is given in figure 1.
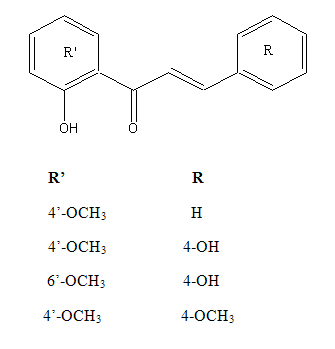
Chemical structure of 2’ hydroxychalcone derivatives5
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
3. Antibacterial properties
The antibacterial activity of chalcones is being increasingly documented. Many research groups either isolated and identified the structure of chalcones that possess antibacterial activity. The bactericidal effects have been related to the ability of α, β unsaturated ketone to undergo a conjugated addition to a nucleophilic group like a thiol group in an essential protein.7
Liquorice (root and rhizome of Glycyrrhiza spp.) is currently used in the tobacco, confectionery, and pharmaceutical industries. Among the retrochalcones (chalcones which do not have an oxygen-function at the 2-position) isolated from Glycyrrhiza inflata licochalcone A and licochalcone C showed potent antibacterial activity especially to Bacillus subtilis,Staphylococcus aureus and Micrococcus luteus. Licochalcone A inhibited the growth of Legionella pneumophila, Legionella longbeacheae, Legionellawadsworthii, Legionella bozemanii, Legionella runoffs and Legionella feelei. Licochalcone A, inhibitory activity was detected against the growth of Helicobacter pyroli in vitro. Licochalcone A was tested against S. aureus and showed that the free hydroxyl group in 4-position of ring B was necessary for the antibacterial activity. On the other hand, no change in its activity is observed when the hydroxyl group in the 4-position of ring A is removed, blocked by a methyl, or replaced by a chlorine. Removal of both hydroxyl groups or blockage of both hydroxyl groups by methylation eliminates the activity completely.8-9
Biological screening of dihydrochalcones showedthat these compounds demonstrated relatively goodactivity against the Gram-positive bacteria S. aureus and B.subtilis, and the Gram-negative bacterium Pseudomonas aeruginosa.10

4. Antileismanial properties
Leishmaniasis is a group of prevalent diseases caused by protozoan parasites belonging to the genus Leishmania. Recently a series of synthetic and naturally occurring chalcone derivatives have been reported to be potential agents against Leishmania in a number of in vitro and in vivo assays.
Chalcones are structurally simple compounds of the flavonoid family and are present in a variety of plant species with a vast spectrum of pharmacological activities including antibacterial, antifungal, immunosuppressive, and antinociceptive properties.11-13 One of the most studied antileishmanial chalcones is licochalcone A isolated from the Glycyrrhiza spp. Chinese plant, which inhibits the parasite enzyme fumarate reductase.14 However, licochalcone A and some synthetic derivatives have also been shown to affect human cell functioning by inhibiting lymphocyte proliferation and cytokine production.15
The selective activity of the 20, 60-dihydroxy-40-methoxychalcone (DMC) isolated from the inflorescences of Piper aduncum against intracellular amastigotes of Leishmania amazonensis and the improvement of its therapeutic activity in mice by encapsulation in biodegradable nanospheres.16-17
Licochalcone A inhibited the activities of both nicotinamide adenine dinucleotide reduced-fumarate reductase (NADH-FRD) and succinate dehydrogenase (SDH) in the permeabilized promastigotes in a concentration-dependent manner. This compound also inhibited the activities of SDH, NADH dehydrogenase (NDH), succinate-cytochrome c reductase (SCC), and NADH-cytochrome c reductase (NCC). The IC50s of licochalcone A for SDH (593 µM), NDH (460 µM), SCC (1,519 µM), and NCC (1,985 µM) after 60 min of incubation were at least 33 times higher than the IC50 for fumarate reductase (FRD) (14 µM). The IC50s of licochalcone A for SDH and NDH in the crude mitochondria of mammalian cells of human peripheral blood mononuclear cells (PBMC) were very high: both were 1.4 µM after 60 min of incubation. The IC50s of licochalcone A for SDH in mammalian cells were more than 67 times higher than the IC50 for FRD in the parasite. Licochalcone A clearly exhibited concentration- and time-dependent inhibitory effects on soluble FRD in the parasite. The IC50 of licochalcone A after 60 min of incubation was 32 µM. Licochalcone A probably first inhibits FRD of the parasite, then influences the parasite respiratory chain and affects the function and ultra structure of the parasite mitochondria, and finally kills the parasite. Licochalcone C inhibited the growth of the L. major parasite to the same extent as licochalcone A.18
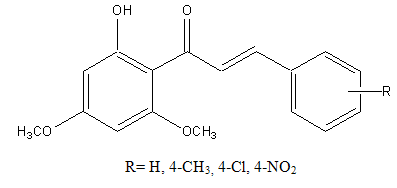
5. Antimalarial properties
Malaria continues to be one of the major public health problems in many tropical countries causing extensive morbidity and loss of life. Annual malaria mortality due to Plasmodium falciparum costs 1-2.7 million lives in Africa alone, comprising of mainly young children. Artemisinin contained in the decoction prepared from the aerial parts of a plant Artemisia annua, used for over thousand years for fever resolution has been re-discovered as the most potent antimalarial drug. Resistance to this drug has not been clinically encountered so far. Chloroquine the only synthetic antimalarial drug which cured malaria for decades, rather than centuries, has fallen to resistance.19 One of the strategies adopted by malariologist to delay the emergence of resistance to artemisinin and its derivatives is to use them in combination with other novel antimalarial(s). Artemisinin based combination therapies (ACTs) instead of artemisinin monotherapy are being advocated and supported by WHO. Currently used ACTs are in demand and by no means the ideal combinations. There is scarcity of artemisinin globally. Thus there is a need to search for novel, inexpensive antiplasmodials as suitable synergistic partners for artemisinin to reduce dependence on artemisinin for malarial treatment.20-22
Chalcone, a biosynthetic product of shikimate pathway, is a class of privileged structure that has a wide range of biological properties. Chalcones are precursors of various flavones and key intermediates for combinatorial assembly of different heterocyclic scaffolds. Chalcone (1, 3-diaryl propenone or 1, 3-diphenyl- 2-propen-1-one) constitutes an important group of natural products, and some of them possess a wide range of biological activities, such as anti-bacterial, anti-fungal, antiviral, anti-inflammatory , anti-tumor, antioxidant, insect anti-feedent, and act as tyrosinase inhibitors. Interest in chalcones as antimalarials was initiated by the discovery of antiplasmodial activity of licochalcone A an oxygenated chalcone isolated from the roots of the Chinese licorice during routine screening. Computational structural analysis also identified chalcones as potential plasmodial cysteine protease inhibitors.23-24
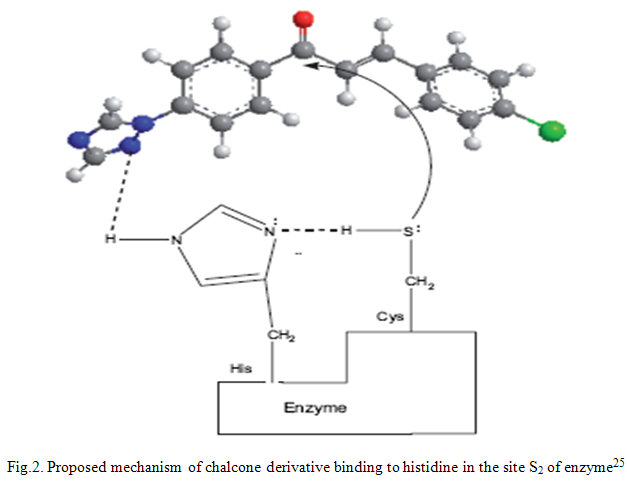
Plasmodium falciparum and Plasmodium vivax are the two major human malaria parasites. P. falciparum is responsible for most deaths, and it has developed resistance to nearly all available drugs. Chalcone derivatives exhibit antimalarial activity by inhibiting plasmodial cysteine protease enzyme. In vitro antiplasmodial results of 4-chloro, 4-methoxy and 3, 4, 5-trimethoxy series suggested that small or medium sized but highly lipophilic groups containing multiple nitrogen or amine in acetophenone moiety impart antiplasmodial potential. Such compounds may provide additional hydrogen bonding with histidine residue present at the active site of the enzyme, cysteine protease. This is the first report in which chalcones containing small highly polar, lipophilic cyclic amines are showing antimalarial potential.25-26 The mechanism of chalcone derivative binding to histidine in the site S2 of enzyme are given in figure 2.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
6. Antifungal properties
Modern medicine has greatly increased the number of immunocompromised patients. Chemotherapy, parenteral nutrition, transplantation surgery and the use of broad-spectrum antimicrobial agents, added to the occurrence of the acquired immunodeficiency syndrome (AIDS), render true ‘living Petri dishes’ individuals, who are highly susceptible to opportunistic infections. Systemic and dermic fungal infections are the cause of great morbi-mortality in this type of patients, being dermatomycoses a serious problem for children of the Third World Nations as a consequence of poor sanitation care.27-28
Since many of the currently available drugs have undesirable side effects, are ineffective against new or reemerging fungi, or develop a rapid resistance, there is an urgent need of a next generation of new antifungal agents, which overcome the above disadvantages. The search is oriented to find new antifungal drugs, which may selectively attack the fungi without inhibiting any biochemical system of the host. Since fungal but not mammalian cells possess a cell wall, its inhibition represents an ideal mode of action for antifungal drugs.29
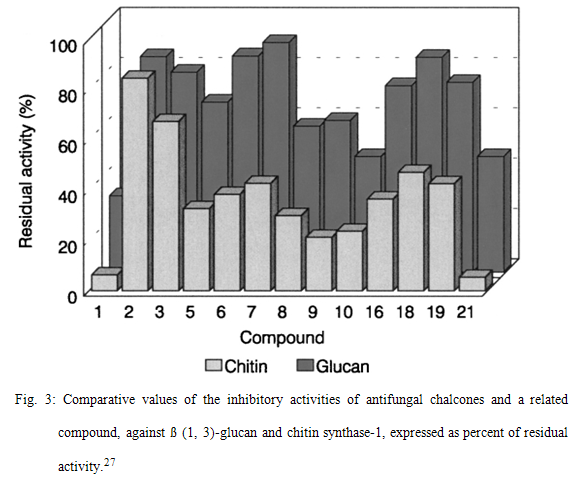
Chalcone derivatives possess moderate antifungal properties.The mechanism of action of most active chalcones was assayed for their capacity of inhibiting in-vitro β (1, 3)-glucan and chitin synthases enzymes that catalyze the biosynthesis of β (1, 3)-glucan and chitin polymers of the fungal cell wall respectively.30Comparative values of the inhibitory activities of antifungal chalcones and a related compound, against β (1, 3)-glucan and chitin synthase-1, expressed as percent of residual activity are given in figure 3.
7. Antidiabetic properties
A new series of chalcone derivatives inhibit aldose reductase enzyme (ALR2) and their specificity towards the target enzyme with respect to other oxidoreductases, such as aldehyde reductase, sorbitol dehydrogenase, and glutathione reductase.
The aldose reductase (ALR2) is the first enzyme of the so-called 'polyol pathway'; in the presence of NADPH it converts glucose to sorbitol, which is further processed by sorbitol dehydrogenase (SD) to fructose. Since ALR2 has low affinity for glucose, flux through this pathway is probably low in most tissues under normoglycemic conditions. On the contrary in diabetic conditions the blood glucose concentration increases sufficiently to provide a substrate for ALR2 in tissues such as nerve, lens, and retina, in which insulin is not necessary for glucose transport across the membrane. The increase glucose flux through the sorbitol pathway and/or the high intracellular accumulation of sorbitol is likely to be involved in the etiology of late-onset diabetic complicance such as neuropathy, nephropathy, retinopathy and cataract.31
Several ALR2 inhibitors have been used to reduce or to delay the development of these diabetic complications, but many problems are associated with this therapy. The first concerns the specificity of the inhibition: ALR2 is a member of the aldo-keto reductase family, a group of enzymes that catalyze the NADPH-dependent reduction of a wide variety of carbonyl compounds, possessing broad and overlapping substrate specificity. The possible consequences arising, in the course of chronic ALR2 inhibitors therapy, from the inhibition of closely related enzymes not involved in the polyol pathway are therefore of legetimate concern in the development of these drugs.. It is also important to determine the selectivity relative to other enzymes such as sorbitol dehydrogenase (SD) and/or glutathione reductase (GR): SD happens to be the second enzyme of the polyol pathway, and its contemporaneous inhibition would give rise to an intracellular accumulation of sorbitol, while the inhibition of GR would cause the accumulation of glutathione disulphide, which is able to modify ALR2, thus rendering it insensitive to the traditional inhibitors.32
Thus, the interesting discovery of the ALR2 inhibitory activity of phenolic compounds like chalcones, devoid of highly acidic carboxylic moiety prompted us to study a series of isoliquiritigenin (2',4,4'-trihydroxychalcone) analogous, taken as a lead compound, containing some modifications on aromatic rings (introduction of methoxy groups, halogen atoms or substitution with heteroaromatic rings) together with the corresponding dihydrochalcone derivatives.33
8. Conclusion
Chalcone is a unique template that is associated with several biological activities. The different pharmacological activities of chalcone derivatives like anti-inflammatory, anti-infective, antidiabetic, antileismanial, antimalarial, antifungal activities etc. have been reported in different reported articles.
References
1. A. O. Fatima, J. Heterocycl. Chem., 41, 327, (2004).
2. M. S. Y. Khan, Indian J. Chem., 42B, 1970, (2003).
3. Z. Nowakowska, Eur. J. Med. Chem., 42, 125, (2007).
4. D. H. Jadhav and C. Ramaa, Indian J. Chem., 46B, 2064, (2007).
5. J. Rojas, M. Paya and M. L. Ferrandiz, Bioorg. Med. Chem. Lett., 12, 1951, (2002).
6. H. S. Ban, K. Suzuki and S. S. Lim, Biochem. Pharmacol., 67, 1549, (2004).
7. D. G. Batt, R. Goodman and J. S. Kerr, J. Med. Chem., 36, 1434, (1993).
8. P. Venturalla, A. Bellino and I. Piozzi, Farmaco. Ed. Sci., 26, 591, (1971).
9. S. S. Tiwari and A. Singh, J. Indian Chem. Soc., 38, 931, (1961).
10. Y. B. Vibhute and M. A. Basser, Indian J. Chem., 42B, 202, (2003).
11. R. Correa, M. A. S. Pereira, D. Buffon, L. Santos, V. F. Cechinel, A. R. S. Santos and R. Nunes, J. Arch. Pharm. Pharm. Med. Chem., 334, 167, (2001).
12. J. R. Dimmock, D. W. Elias, M. A. Beazely and N. M. Kandopu, Curr. Med. Chem., 6, 125, (1999).
13. J. C. Bail, C. Pouget, C. Fagnere, J. Basly, A. Chulia and G. Habriouse, Life Sci., 68, 751, (2001).
14. M. Chen, L. Zhai, S. B. Christensen, T. Theander, A. Kharazmi, Antimicrob. Agents Chemoth., 45, 2023, (2001).
15. L. Barfod, K. Kemp, M. Hansen and A. Kharazmi, Int. Immunopharmacol., 2, 545, (2002).
16. E. C. Torres-Santos, D. Moreira, M. A. Kaplan and B. Rossi- Bergmann, Antimicrob. Agents Chemother., 43, 1234, (1999).
17. E. C. Torres-Santos, J. M. Rodrigues, D. L. Moreira, M. A. Kaplan and B. Rossi-Bergmann, Antimicrob. Agents Chemother., 43, 1776, (1999).
18. P. Boeck, P. C. Leal and R. A. Yunes, Bioorg. Med. Chem., 14, 1538, (2006).
19. N. Mishra, P. Arora and B. Kumar, Eur. J. Med. Chem., 43, 1530, (2008).
20. J. R. Vishnu, S. S. Abhishek, S. Shalini and C. Subhash, Bioorg. Med. Chem. Lett., 10, 2159, (2000).
21. J. May and C. G. Meyer, Trends Parasitol., 19, 432, (2003).
22. WHO ed, WHO Guidelines for the Treatment of Malaria, WHO, p.1086 (1998).
23. V. K. Bhasin and L. Nair, Lancet Infect. Dis., 3, 609, (2003).
24. M. Chen, T. G. Theander, S. B. Christensen, L. Hviid, L. Zhai and A. Kharazmi, Antimicrob. Agents Chemother., 38, 1470, (1994).
25. S. Khatib, O. Nerya, R. Musa, M. Shmuel, S. Tamir and J. Vaya, Bioorg. Med. Chem., 13, 433, (2005).
26. K. R. Yerra, S. Fang and Y. M. Tzeng, Bioorg. Med. Chem., 12, 2679, (2004).
27. S. N. Lopez, M. V. Castelli and S. A. Zacchino, Bioorg. Med. Chem., 9, 1999, (2001).
28. V. J. Cid, A. Duran, R. F. Del, M. P. Snyder, C. Nombela and M. Sanchez, Microbiol. Rev., 345, (1995).
29. F. M. Klis, Yeast., 10, 4381, (1994).
30. T. C. White, K. A. Marr and R. A. Bowdenn, Clin. Microbiol. Rev., 11, 382, (1998).
31.F. Severi, S. Benevenuti and L. Costantino, Eur. J. Med. Chem. 33, 859, (1998).
32. P. F. Kador, Med. Res., 8, 325, (1988).
33. D. R. Tomlinson, E. J. Stevens and L. T. Diemel, Trends Pharmacol. Sci., 293, (1994).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






