

Using data generated from patients and mice with genetic mutation for the disorder Usher syndrome, researchers from the University of Maryland School of Medicine (UMSOM), the National Institutes of Health’s National Eye Institute (NEI), and National Institute on Deafness and Other Communication Disorders (NIDCD), documented the natural history of vision impairment in patients and identified the cell mechanism behind progressive vision loss.
Based on these findings, published on November 9, 2021, in the journal eLife, the team was able to test a retinoid therapy that improved vision in mice with Usher syndrome. The researchers said assessing a similar therapy should now be considered in people with Usher syndrome to see if this therapy might slow vision loss.
Usher syndrome type 1F (USH1F) causes deafness, progressive vision loss, and balance issues. Among Ashkenazi Jews, there is a 2% chance each person is a carrier of the Usher syndrome type 1F mutation, accounting for approximately 60% of their Usher syndrome type 1 cases. There are no approved therapies to prevent vision loss or restore vision in people with Usher syndrome.
“The drug we used in mice may provide a first step to improve eye health in people with Usher syndrome type 1F,” said Zubair M. Ahmed, PhD, Professor of Otorhinolaryngology–Head & Neck Surgery and Ophthalmology at UMSOM. “Unfortunately, these drugs will not permanently cure loss of vision, as the drug does not repair damage or prevent degeneration of the eyes. However, it should improve the function of the tissue that these patients still have.”
First author of the study Saumil Sethna, PhD, Instructor in Otorhinolaryngology–Head & Neck Surgery, said, “There are currently FDA-approved relatives of these retinoid drugs that are available and have passed clinical trials for safety, along with others that are in phase II clinical trials to treat other types of vision loss disorders.”
The team hopes to partner with one of the companies testing these drugs to launch a clinical trial in patients with Usher syndrome type 1F to see if it can help by preventing continuing vision loss.
“The identification of a key mutation in the PCDH15 gene nearly two decades ago was a critical breakthrough, facilitating the diagnosis of and carrier screening for a certain form of Usher syndrome, now resulting in the discovery of a potential preventative therapy for vision loss associated with the syndrome,” said Thomas B. Friedman, Ph.D., Chief of the Laboratory of Molecular Genetics at the NIDCD. “This work exemplifies the value of basic science research in driving the development of novel diagnostics and therapeutics.”
Prior to this study, only anecdotal data had been reported for Usher syndrome type 1F without any detailed data analysis of the worsening eye abnormalities over time. In the early 2000s, co-authors on this study, including Dr. Friedman at NIDCD, initiated an Usher syndrome natural history project and enrolled 13 study participants with Usher syndrome 1F to follow the natural progression of their accompanying blindness over 20 or more years. Dr. Ahmed was a Postdoctoral Fellow in Dr. Friedman’s laboratory at the time. Wadih Zein, MD, an NEI Ophthalmologist with expertise in inherited retinal degenerations, served as principal investigator on the natural history study and summarized patient findings collected over the years. Based on analysis of vision tests, Dr. Zein found that the Usher syndrome type 1F mutation led to 50% of the study participants being legally blind by age 50.
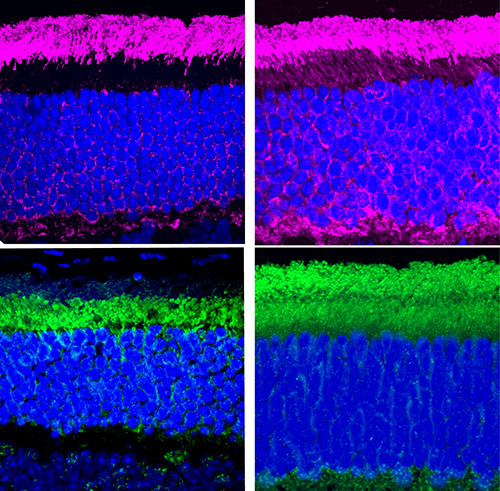
In eye tissue from an Usher syndrome mouse model (right), the light-dark cycle proteins (purple and green) are spread throughout the two photoreceptor compartments rather than separated in one or the other as in the healthy mouse eye tissue (left).
Separately, Dr. Ahmed created a mouse with an Usher syndrome found in 13 of Dr. Friedman’s patients. This mutation in the gene PCDH15 leads to a shortened version of the protein protocadherin-15. However, the mechanism for how this mutant protocadherin-15 led to blindness was unknown. To unravel this, Dr. Friedman’s Dr. Zein’s, and Dr. Ahmed’s teams decided to collaborate and compare the eye characteristics from humans and mice with this genetic condition that they had independently collected over the years.
Comparing the Usher syndrome mouse model to healthy mice, Dr. Ahmed’s team identified two functions of protocadherin-15. First, protocadherin-15 helps light-dark cycle proteins move back and forth between the different compartments of the light-detecting photoreceptors in the eye. Second, protocadherin-15 is also required for recycling molecules essential for functioning eye tissue, known as retinoids. The Usher syndrome type 1F mutant mice had reduced levels of retinoids in a certain kind of eye cell.
Next, the researchers gave a retinoid drug to the Usher syndrome mice to see if it improved their vision. Retinoid injections into the Usher syndrome mice increased the electrical activity in the eyes of both younger and adult mice, indicating improved vision function.
“Aside from retinoid replacement, we can also think about developing more permanent therapies to treat or prevent blindness in people with Usher syndrome type 1F that may correct or replace the other functions of protocadherin-15, as well,” said Dr. Ahmed.
E. Albert Reece, MD, PhD, MBA, Executive Vice President for Medical Affairs, UM Baltimore, and the John Z. and Akiko K. Bowers Distinguished Professor and Dean, University of Maryland School of Medicine said, “People born with deafness, like individuals with Usher syndrome, rely on their other senses to communicate with and understand the world. Subsequent vision loss can pose significant challenges and can be isolating. This work here builds foundational discoveries that may one day improve the lives of people with rare genetic disorders such as Usher syndrome.”
Other authors on the study include Research Fellow Sehar Riaz, MS; Arnaud Giese, PhD, Instructor in Otorhinolaryngology-Head & Neck Surgery; and Saima Riazuddin, PhD, MPH, MBA, Professor of Otorhinolaryngology-Head & Neck Surgery, all at UMSOM; Robert Hufnagel, MD, PhD, Chief, Medical Genetics and Ophthalmic Genomics Unit; Todd Duncan, MS, Senior Research Assistant, and T. Michael Redmond, PhD, Chief, both of Molecular Mechanisms Section of the NEI; and Carmen Brewer, PhD, Chief Research Audiologist; and Andrew Griffith, MD, PhD, Ex Scientific Director of NIDCD; and Julie Schultz, PhD, previously at the NIDCD, is now affiliated with GeneDx, Inc.











