

 About Authors:
About Authors:
Tarun Patel , Prof. Dr. Vipin Kukkar, Nilesh Sovasia
Seth G.L. Bihani S.D. College of Technical Education,
Institute of Pharmaceutical Sciences and Drug Research,
Sri Ganganagar, Rajasthan, INDIA
INTRODUCTION
When we speak of a molecule as being raised to a higher electronic level we mean that an electron has been changed from one orbital to another orbital of higher energy. This electron can be of any kinds we have encountered –a σelectron, a π electron or an n electron. In Ultraviolet region we are confined only to the excitation of the comparatively loosely held n and πelectrons.2
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1249
IMPORTANT IN THIS ARTICLE:
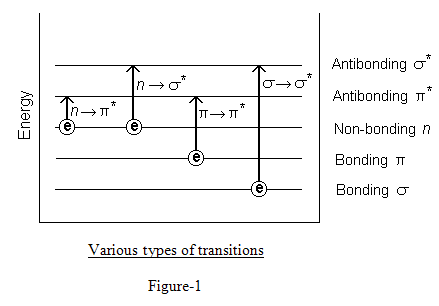
Out of the above mentioned transitions only n→ π* and π→ π* are of use to the analytical chemist working on the ultraviolet spectrophotometer.
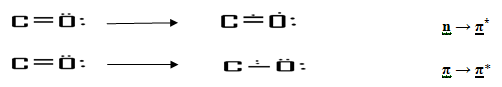
When light – either visible or ultraviolet – is absorbed by valence (outer) electrons. These Electrons are promoted from their normal (ground) states to higher energy (excited) states .The energies of the orbitals involved in electronic transitions have fixed values. Because energy is quantised, It seems safe to assume that absorption peaks in a UV/visible spectrum will be sharp peaks. However, this is rarely, if ever, observed. Instead the spectrum has broad peaks .This is because there are also vibrational and rotational energy levels available to absorbing materials.
Ultraviolet- visible spectroscopyor ultraviolet - visible spectrophotometery (UV-Vis or UV/Vis) involves the spectroscopy of photons in the UV-visible region.
There is an interaction between UV visible light and sample which is in solution form. As a result of this interaction some photons (photons of UV-Vis EMR) are absorbed and this absorption of UV visible is measured by an instrument named UV visible spectrophotometer.
UV visible is low energy EMR hence generally no ionization is take place but electronic transition of lone pair and π electron take place (200-800 nm).6
QUANTUM MECHANICS
Quantum mechanics (QM) is a set of scientific principles describing the known behavior of energy and matter that predominate at the atomic and subatomic scales. QM gets its name from the notion of a quantum, and that quantum value is the Planck constant. The wave–particle duality of energy and matter at the atomic scale provides a unified view of the behavior of particles such as photons and electrons. While the notion of the photon as a quantum of light energy is commonly understood as a particle of light that has an energy value governed by the Planck constant, what is quantized for an electron is the angular momentum it can have as it is bound in an atomic orbital.Electrons have certain properties of particles and certain properties of waves. Electrons have mass and charge like particles. Because they are so small and are moving so fast, electrons have no defined position. Their location is best described by wave mechanics (i.e. a three dimensional wave) and a wave equation called the Schrödinger equation. Solutions of the Schrödinger equation are called wave functions and are represented by the Greek letter psi. Each wave function describes a different orbital. There are many solutions to the Schrödinger equation for a given atom.
Atomic orbitals:- The region in space where an electron is likely to be found called an orbital. There are different kinds of orbitals, which have different sizes and different shapes, and which are disposed about the nucleus in specific ways. The particular kind of orbital that an electron occupies depends upon the energy of the electron.5
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
CONCEPT OF MOLECULAR ORBITALS
Theory of molecular orbitals comes under the preview of quantum mechanism. Erwin schrodinger formulated a wave equation which has a series of solutions called wave functions, each corresponding to a different energy level for the electron.
In molecules, as an isolated atoms, electrons occupy orbitals and in accordance with much the same “rules”. These molecular orbtials are considered to be centered about many nuclei, perhaps covering the entire molecule; the distribution of nuclei and electrons is simply the one that results in the most stable molecule.
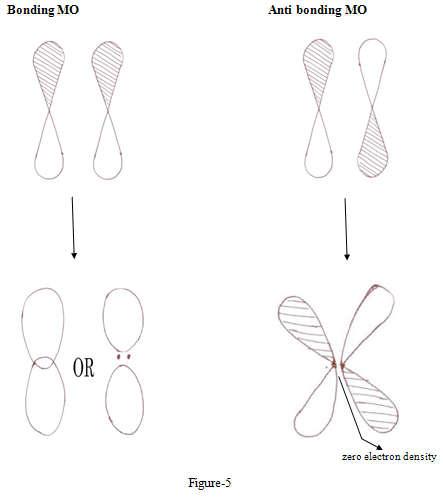
Constructive interference of two wave function leads to high probability density between the nuclei, leading to bonding orbital while in antibonding MOs wave function interfare destructively leading to low electron density that is greater repulsions
Thus electrons in a bonding MOs tend to hold the atoms together and electrons in antibonding tend to force atoms apart.8
Linear Combination of Atomic Orbitals [LCAO]
As we know that the number of formed molecular orbitals is equal to the number of component atomic orbitals. AOs may overlap either axially or collaterally. This can be understood diagrammatically as shown below
Axial Overlapping
Axial Overlapping
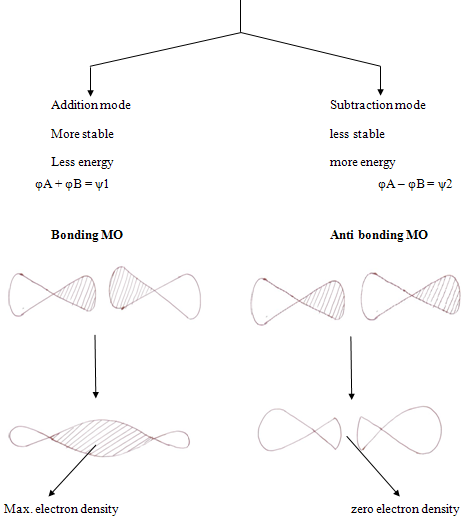
Figure 3
Co-lateral overlapping
lateral Overlaping
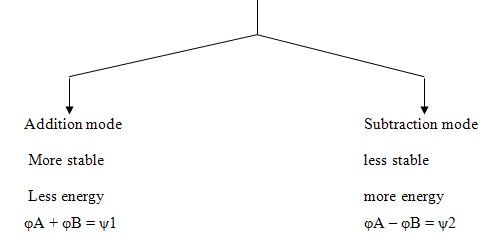
ENERGY LEVELS OF BONDING AND ANTI-BONDING MOLECULAR ORBITALS
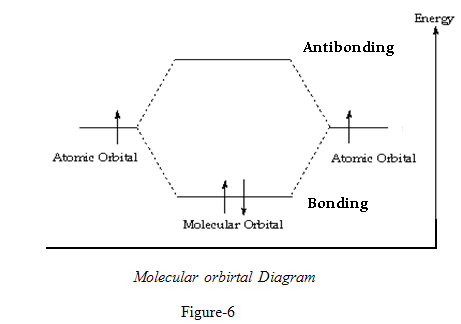
The two P atomic orbitals combine to form two molecular orbitals, one bonding and one antibonding .5
ELECTRONIC CONFIGURATION OF SOME MOLECULES
Ethylene configuration of π electrons in the ground state and the excited state
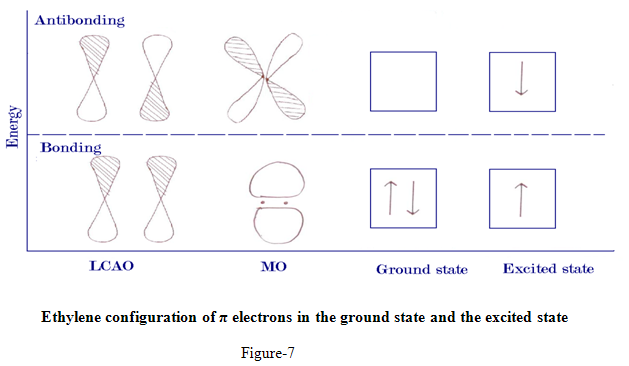
For the π electrons of ethylene, there are two molecular orbitals since there are two linear combinations of the two component p orbitals. The broken line in the figure indicates the non-bonding energy levels. Below it lies the bonding orbitals, π and above it lies the antibonding orbitals π*. Normally a molecule exists in the state of lowest energy, the ground state. But absorption of light of the right frequency in the ultraviolet region rises a molecule to an excited state, a state of higher energy, in the ground state of ethylene both πelectrons are in the π orbital, this configuration is specified as π2, where the superscript tells the no. of electrons in the orbital. In the excited state one electron is in the πorbital and the other still of opposite spin- is in the π*orbital, this configuration, ππ*is naturally the less stable since only one electron helps to hold the atoms together, while other tend to force them apart.
1,3- butadiene .configuration of π electrons in the ground state and the first excited state
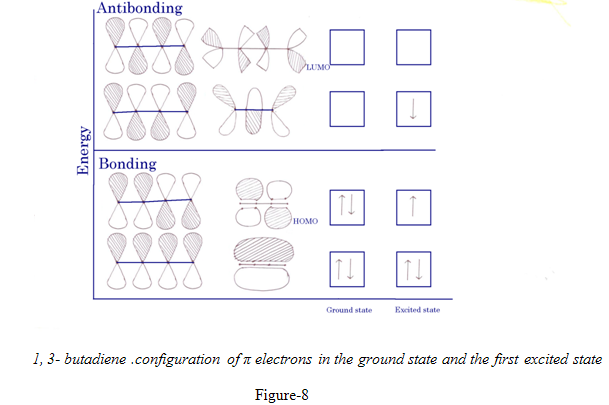
For 1,3-butadiene,with 4 component p orbitals, there are 4 MO for π electrons. The ground state has the configuration ψ12 ψ22, i.e. there are two electrons in each of the bonding orbitals ψ1 & ψ2.the higher of these ψ2, resembles two isolated π orbitals, although it is of somewhat lower energy, orbital ψ1 encopasses all four carbons, this delocalization provides the net conjugated system. Absorption of light of the right frequency rises one electron to ψ3
hv
ψ12 ψ22 ---------------> ψ12 ψ2 ψ3
ground state lowest excited state
HOMO refers to Highest Occupied Molecular Orbital
LUMO refers to Lowest Unoccupied Molecular Orbital
The two orbitals best matched in energy will be the highest occupied molecular orbitals or HOMO. on one reactant, and the lowest energy unoccupied MO,or LUMO on the other.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
POSSIBLE HOMO-LUMO COMBINATIONS
Table-1
|
HOMO |
LUMO |
RESULT |
|
An occupied n orbital |
An empty n orbital |
Bond formation only |
|
An occupied n orbital |
A σ*orbital |
Bond formation and bond rupture |
|
An occupied n orbital |
A π*orbital |
Bond formation and bond rupture |
|
A πorbital |
An empty orbital |
Bond formation and bond rupture |
|
A π orbital |
A σ*orbital |
Bond formation and bond rupture |
|
A π orbital |
A π* orbital |
Bond formation and bond rupture |
|
A σ orbital |
An empty orbital |
Bond formation and bond rupture |
|
A σ orbital |
A σ* orbital |
Bond formation and bond rupture |
|
A σ orbital |
A π* orbital |
Bond formation and bond rupture |
Transition probability
It is not essential that exposure of a compound to ultraviolet or visible light must always gives to an electronic transition. On the other hand, the probability of a particular electronic transition has found to depend €d upon the value of molar extinction coefficient and certain other factors. According transitions can be divided into two categories.
(i) Allowed transitions
(ii) Forbidden transitions
(i) Allowed transitions – these are transitions having molar coefficient 104 or more. These are generally designated as π→ π transitions. For example in 1,3-butadiene which exhibits absorption at 217nm has €max value 21000 represents an allowed transition. These transition are mainly favoured due to symmetry relationship. For e.g.
1,3- butadiene absorbs at 217nm and has molar absorptivity of 21000
(ii) Forbidden transitions – these are transitions for which €max is generally less than 104 . for example transition of saturated aldehyde showing weak absorption near 290nm and having €max 100 has been a forbidden transition. For e.g.
Carbonyl group absorbs at 300nm and a molar absorptivity of 10-100.5
TRANSITIONS IN ULTRAVIOLET SPECTROSCCOPY
Electronic transitions in UV-visible spectroscopy which are important are
n→ π* & π→ π*transitions.
(a) n→ π* transitions. - In this transition, an electon of unshared electron pair on a hetero atom is excited to π* antibonding orbital. This transition involves least amount of energy than all the transitions and therefore, this transition gives rise to an absorption band at longer wavelengths. In saturated aliphatic ketones, e.g., the n→ π*transitions around 280 nm is the lowest energy transitions. This n→ π* transitions is “forbidden” by symmetry considerations, thus the intensity of the band due to this transition is low, although the wavelength is long (lower energy).
(b) π→ π* transitions - This transition is available in compounds with unsaturated centres, e.g., simple alkenes, aromatics, carbonyl compounds, etc. this transition requires lesser energy then transition in a simple alkene, although several transitions are available , the lowest energy transition is the π→ π*transition and a absorption band around 170nm-190nm in unconjugated alkenes is due to this transition in the case of , e.g., saturated ketones, the most intense band around 150nm is due to π→ π*transition.7
DESIGNATION OF BANDS
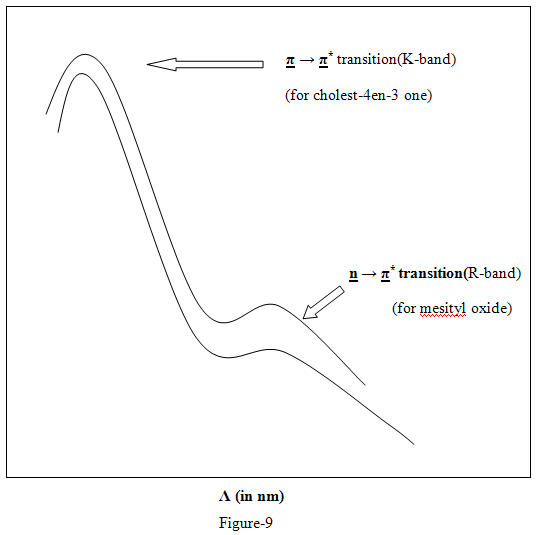
K- Band
One may designate the UV absorption bands by using electronic transitions or the letter designation. The band due to π→ π* transitions in a compound with conjugated π system is usually intense (€max.>10000) and is frequently referred to as the k-band (german- konjugierte). The examples of the compounds in which k-band appears are butadiene, Mesityl oxide. Benzene itself displays three absorption bands at 184,204 and 256nm and of these the band at 204nm is often designated as k-band, and this used in other benzenes as well.
Eg. Conjugated diene, triene, polyene, enones and aromatic rings
R- Band
The n→π* transition (R-band german radikalartig) in compounds with single chromatographic groups i.e., carbonyl or nitro are forbidden with € value less than 100.
In conjugated systems the energy separation between the ground and excited states is reduced and the system then absorbs at longer wavelengths and with a greatly increased intensity (k-band is intense and at longer wavelength). Moreover, due to the lessening of the energy gap, the n→ π* transition due to the presence of the heteroatom and lone pair i.e. the r-band also undergoes a red shift with little change in intensity.
Eg. Acetone, acroline, methyl vinyl ketone, acet aldehyde, acetophenone, croton aldehyde
B-Band
These bands are observed in aromatic compounds and hetero aromatic compounds. Here B refers to Benzenoid bands
Eg. Benzene, tolune, acetophenone, benzoic acid, napthelene, styrene
E- Bands
Such band originate due to electronic transition in the benzenoid system of the ethylinic part enclosed in cyclic conjugation. Here E refers to Ethylinic. These are further classified as E1 and E2
Eg. Benzene, nepthelene, anthracene, quinolene7
DIFFERENT EFFECTS
1. Effect of solvent
The transitions of polar bonds, like c=o but not ethylene, are affected by solvent polarity as solvent polarity is increased, π→ π*bands undergo red shifts. This is so since excited state is more polar than the ground state and hence stabilization is greater relative to the ground state with two n electrons receives greater stabilization than the excited state with only n electron. These opposite trends are clear by examining the data of mesityl oxide. There is more on shift of bands with solvents.
2. Effect of conjugation
Absorption in near UV that is above 200 nm is invariably associated with the presence of unsaturated groups or atoms with unshared pairs of electrons the saturated hydrocarbon which do not have these structural elements observe below 200nm reason, not of much significance for structural study of organic compounds. Thus interstically a complex steroid molecule cholest-4-ene-3 one is easily recognized to have an α-ß unsaturated keton moiety, similar to that in mesityl oxide by their spectral resemblance.5
3. Effect of PH
In alkaline PH π → π* transition is more favoured and in acidic PH n → π* transition is more favoured. For eg.
(1) P- nitrophenol ---> p- nitrophenol (+M effect increased)
Alkaline PH
π → π* transition more favoured
less energy is required
absorption would take place at longer wavelength
(2) p-amino phenol ---> P-amino phenol
Acidic PH
n → π* transition is more favoured
More energy required
Blue shift
Absorption at shorter wavelength
REFERENCES
1. Carry F.A., “organic chemistry”, fourth ed., new York, McGraw hill.
2. Chatwal gurdeep R. and Anand sham K., “instrumental methods of chemical analysis”, edited by M. Arora, published by Himalaya publishing house, new delhi, 2005, page no.-2.150-2.154
3. Dr. Shankar ravi, “textbook of pharmaceutical analysis”, third edition, Rx publication, page no.-2.2-2.5
4. Jag Mohan, “organic analytical chemistry theory and practice”, published by Narosa publication house, new delhi. Page no.-347-350.
5.Kalsi P.S., “spectroscopy of organic compound”, sixth edition, new delhi. New age international Pvt. Ltd.
6.Medham J, Denny R.C., Barner J.D., Thomas M. “vogel’s textbook of qualitative chemical analysis”, sixth edition, London. Pearson education Ltd., 2008.
7.Sharma Y.R. “elementary organic spectroscopy”, page no.-15-19
8.Willard H.H., Meritt L.L., Dean J.A., Settle F.A. “instrumental method of analysis” 7th edition, India: CBS publishers and distributors.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






