

 ABOUT AUTHORS:
ABOUT AUTHORS:
Rajani Pathuri*, M.Muthukumaran, B.Krishnamoorthy, Amreen Nishat
Montessori Siva Sivani Institute of Science & Technology-College of pharmacy
Mylavaram, Vijayawada, Andhrapradesh-521230
*rajani.prakash4@gmail.com
ABSTRACT
Analytical methods development and validation play important roles in the discovery, development and Manufacture of pharmaceuticals. Method development is the process of proving that an analytical method is acceptable for use to measure the concentration of an API in a specific compounded dosage form which allow simplified procedures to be employed to verify that an analysis procedure, accurately and consistently will deliver a reliable measurement of an active ingredient in a compounded preparation. The analytical method validation is essential for analytical method development and tested extensively for specificity, linearity, accuracy, precision, range, detection limit, quantization limit, and robustness. In summary, analytical method development and validation allows to confirm that an accurate and reliable potency measurement of a pharmaceutical preparation can be performed.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-1691
INTRODUCTION
The number of drugs introduced into the market is increasing every year. These drugs may be either new entities or partial structural modification of the existing one. Very often there is a time lag from the date of introduction of a drug into the market to the date of its inclusion in pharmacopoeias. This happens because of the possible uncertainties in the continuous and wider usage of these drugs, reports of new toxicities (resulting in their withdrawal from the market), development of patient resistance and introduction of better drugs by competitors. Under these conditions, standards and analytical procedures for these drugs may not be available in the pharmacopoeias. It becomes necessary, therefore to develop newer analytical methods for such drugs.[1-5]
ANALYTICAL METHOD DEVELOPMENT
Analytical chemistry deals with methods for identification, separation, and quantification of the chemical components of natural and artificial materials. [6] The choice of analytical methodology is based on many considerations, such as: chemical properties of the analyte and its concentration sample matrix, the speed and cost of the analysis, type of measurements i.e., quantitative or qualitative and the number of samples. A qualitative method yields information of the chemical identity of the species in the sample. A quantitative method provides numerical information regarding the relative amounts of one or more of the analytes in the sample.
The steps of method development and method validation the steps of method development and method validation
-Method development plan definition
-Background information gathering
-Laboratory method development, it includes various stages namely sample preparation, specific analytical method, detection and data processing
-Generation of test procedure
-A well-developed method should be easy to validate. A method should be developed with the goal to rapidly test preclinical samples, formulation prototypes, and commercial samples. There are five common types of analytical methods, each with its own set of validation requirements
-Identification tests
-Potency assays
-Quantitative tests for impurities
- Limit test for the control of impurities
-Specific tests
The first four tests are universal tests, but the specific tests such as particle-size analysis and X ray diffraction are used to control specific properties of the active pharmaceutical ingredient (API) or the drug product. [7-8]
The most widely used methods for quantitative determination of drugs and metabolites in biological matrices such as blood, serum, plasma, or urine includes Gas chromatography ,(GC) High-performance liquid chromatography, (HPLC) [9-10] Thin layer chromatography, (TLC) combined GC and LC mass spectrometric (MS) procedures such as LC-MS [11-12] LC-MS-MS, [13-14] GC-MS, [15-16] and GC-MSMS, techniques like NMR is used for structure identification.
Chromatography in different forms is the leading analytical method for separation of components in a mixture. The chromatographic procedure for the separation of substances is based on differences in rates of migration through the column arising from different partition of the compounds between a stationary phase (column packing) and a mobile phase transported through the system. [17] Chromatographic methods can be classified according to the physical state of the mobile phase into the following basic categories: gas chromatography ,(GC) supercritical fluid chromatography (SFC) and liquid chromatography (LC). The technique was originally developed by the Russian botanist M.S. Tswett in 1903. [18-19]
[adsense:468x15:2204050025]
Today TLC is rapidly becoming a routine analytical technique due to its advantages of low operating costs, high sample throughput and the need for minimum sample preparation. The major advantage of TLC is that several samples can be run simultaneously using a small quantity of mobile phase unlike HPLC thus reducing the analysis time and cost per analysis .[20-21] An enhanced form of thin layer chromatography (TLC) is called as High performance thin layer chromatography (HPTLC). [22-23] A number of enhancements can be made to the basic method of thin layer chromatography to automate the different steps, to increase the resolution achieved and to allow more accurate quantitative measurements
Liquid chromatography can be categorized on the basis of the mechanism of interaction of the solute with the stationary phase as: adsorption chromatography ,(liquid-solid chromatography) partition chromatography, (liquid-liquid chromatography) ion-exchange chromatography ,(IEC) size exclusion chromatography (SEC) and affinity chromatography
Early work in liquid chromatography was based on highly polar stationary phases, and nonpolar solvents served as mobile phases, this type of chromatography is now referred to normal-phase liquid chromatography (NPLC).[24] Chromatography on bare silica is an example of normal-phase chromatography. In reversed-phase high performance liquid chromatography ,(RP-HPLC) the stationary phase is nonpolar, [25-26] often a hydrocarbon, and the mobile phase is relatively polar. [27] In RP-HPLC, the most polar component is eluted first, because it is relatively most soluble in the mobile phase.
The definite break-through for liquid chromatography of low molecular weight compounds was the introduction of chemically modified small diameter particles (3 to 10μm) e.g., octadecyl groups bound to silica in the late 1960s. The new technique became rapidly a powerful separation technique and is today called high performance liquid chromatography (HPLC).
HPLC-UV diode-array detection (DAD) [28-29] and HPLC-MS techniques take advantage ofchromatography as a separation method and DAD or MS as identification and quantification methods. The HPLC equipment consists of a high-pressure solvent delivery system, a sample auto injector, a separation column, a detector (UV or DAD) a computer to control the system and display results.
Ultra performance liquid chromatography (UPLC) is a recent technique in liquid chromatography, which enables significant reductions in separation time, solvent consumption and analysis time as compared to the conventional HPLC. [30-31]
Sample preparation
The purpose of sample preparation is to create a processed sample that leads to better analytical results compared with the initial sample. The prepared sample should be an aliquot relatively free of interferences that is compatible with the HPLC method and that will not damage the column .[32] The main sample preparation techniques are liquid-liquid extraction (LLE) [33-34] and solid-phase extraction (SPE). [35] In these methods the analyte of interest was separated from sample matrix, so that as few potentially interfering species as possible are carried through to the analytical separation stage.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Detection
After the chromatographic separation, the analyte of interest is detected by using suitable detectors. Some commercial detectors used in LC are: ultraviolet (UV) detectors ,[36] fluorescence detectors, electrochemical detectors, refractive index (RI) detectors and mass spectrometry (MS) detectors. The choice of detector depends on the sample and the purpose of the analysis.
The UV detectors are the most common HPLC detectors since they are robust, cheap, easy to handle, and since many solutes absorb light in this frequency range. [37] The ordinary UV detector measures the absorbance at one single wavelength at the time. A diode-array detector (DAD) can measure several wavelengths at the same time, and since no parts are moved to change wavelength or to scan, there are no mechanical errors or drift with time.
DAD detectors have been proposed for various applications, such as preliminary identification of a steroidal glycoside in seed,[38] peptide mapping ,[39] assay of sulfamethazine in animal tissues, [40] or identification of pesticides in human biological fluids. [41]
HPLC with a mass spectrometer detector (LC-MS)[42-43] showed superior sensitivity and selectivity compared to HPLC-UV methods. [44]
Table No.1: Use of analytical methods - generics
|
CLINICAL |
PHARMACEUTICAL |
METHODS |
||
|
At Initial Phase of pharmaceutical Development |
||||
|
To determine bioavailability in healthy volunteers |
To develop a stable and reproducible formulation for the manufacture of bioequivalence, dissolution, stability and pilot-scale validation batches |
To understand the profile of related substances and to study stability, To start measuring the impact of Key product and manufacturing process parameters on consistent FPP quality |
||
|
At advanced phase of pharmaceutical development |
||||
|
To prove bioequivalence after critical variations to the prequalified dossier |
To optimize, scale-up and transfer a stable and controlled manufacturing process for the prequalification product |
To be robust, transferable, accurate and precise for specification setting, stability assessment and QC release of prequalified product batches |
||
Figure 1 Prerequisites for analytical method validation
Table No 2 Method development life cycles

Analytical Method validation
Successful acceptance of the validation parameters and performance criteria, by all parties involved, requires the cooperative efforts of several departments, including analytical development, QC, regulatory affairs and the individuals requiring the analytical data. The operating procedure or the Validation Master Plan (VMP) should clearly define the roles and responsibilities of each department involved in the validation of analytical methods.[45]
* Quality control plan and implementation for routine
For any method that will be used for routine analysis, a QC plan should be developed. This Plan should ensure that the method, together with the equipment, delivers consistently
Accurate results. The plan may include recommendations for the following:
· Selection, handling and testing of QC standards
· Type and frequency of equipment checks and calibrations (for example, should the wavelength accuracy and the baseline noise of an HPLC UV detector be checked after each sample analysis, or on a daily or weekly basis?)
· Type and frequency of system suitability testing (for example, at which point during the sequence system should suitability standards be analyzed?)
· Type and frequency of QC samples (for example, should a QC sample be analyzed after 1, 5, 20 or 50 unknown samples, and should there be single or duplicate QC sample Analysis, or should this be run at one or several concentrations?)Acceptance criteria for equipment checks, system suitability tests and QC sample Analysis Action plans in case criteria 2, 3 and/or 4 are not met.
Validated routine methods are transferred between laboratories at the same or different sites when contract laboratories offer services for routine analysis in different areas or when products are manufactured in different areas. When validated routine methods are transferred between laboratories and sites, their validated state should be maintained to ensure the same reliable results in the receiving laboratory. This means the competence of the receiving laboratory to use the method should be demonstrated through tests, for example, repeat critical method validation experiments and run samples in parallel in the transferring and receiving laboratories. The transfer should be controlled by a procedure, most likely some method parameters have to be changed or adjusted during the life of the method if the method performance criteria fall outside their acceptance criteria. The question is whether such change requires revalidation. In order to clarify this question upfront, operating range should be defined for each method, either based on experience with similar methods or else investigated during method development. These ranges should be verified during method validation in robustness studies and should be part of the method characteristics. Availability of such operating ranges makes it easier to decide when ultra method should be revalidated. A revalidation is necessary whenever a method is changed, and the new parameter lies outside the operating range. If, for example, the operating range of the column temperature has been specified to be between 30 and 40°C, the method should be revalidated if, for whatever reason, the new operating parameter is 41°C. Revalidation is also required if the scope of the method has been changed or extended, for example, if the sample matrix changes or if operating conditions change. Furthermore, revalidation is necessary if the intention is to use instruments with different characteristics, and these new characteristics have not been covered by the initial validation. For example, an HPLC method may have been developed and validated on a pump with a delay volume of 5 mL, but the new pump has a delay volume of only 0.5 mL.Whenever there is a change that may require part or full revalidation, the change should follow a documented change control system. The change should be defined, authorized for Implementation and documented.[46-47]
Possible changes may include
* New samples with new compounds or new matrices,
* New analysts with different skills,
* New instruments with different characteristics,
* New location with different environmental conditions,
* New chemicals and/or reference standards and
* Modification of analytical parameters.
When should methods be validated?
A method should be validated when it is necessary to verify that its performance parameters are adequate for use for a particular analytical problem. For example:
· Method just developed
· Revised method or established method adapted to a new problem;
· When a review of quality control indicates an established method is changing with time;
· When an established method is used in a different laboratory, with different analysts or with different equipment
· Demonstration of the equivalence between two methods, e.g. a new method and a Standard. Certain areas of analytical practices, such as in clinical chemistry will specify Validation requirements relevant to the method. This ensures that particular validation Terminology together with the statistics used is interpreted in a manner consistent within the relevant sector. Official recognition of a method may require characterization using a collaborative study.
Parameters for method validation
The parameters for method validation have been defined in different working groups of National and international committees and are described in the literature. Unfortunately, some of the definitions vary between the different organizations. An attempt at harmonization was made for pharmaceutical applications through the ICH where representatives from the Industry and regulatory agencies from the United States, Europe and Japan defined Parameters, requirements and, to some extent, methodology for analytical methods validation[48-49]
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
The various parameters are:
1. Selectivity/Specificity
2. Precision and Reproducibility
3. Accuracy and Recovery
4. Stability
5. Range
6. Limit of Detection
7. Limit of Quantization
8. Repeatability
9. Reproducibility
10. Measurement Uncertainty
11. Sensitivity
12. Ruggedness
1. Selectivity/specificity
The terms selectivity and specificity are often used interchangeably, the term specific generally refers to a method that produces a response for a single analyte.Another aspect of selectivity which must be considered is where an analyte may exist in the sample in more than one form such as free or complexes; inorganic or organo metallic; or the possibility of a component such as Chromium ion being present in different oxidation states such as Cr3+ or Cr6+.Precision and reproducibility Precision is method and concentration specific, which in practice can be very varied. The two most common precision measures are ‘repeatability’ and reproducibility’.[50] The term ‘set’ is defined as referring to a number (n) of independent replicate measurements of some property. Standard deviation is the square root of the sum of squares of deviations of individual results for the mean, divided by one less than the number of results in the set. The standard deviation S, is given by
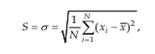
Standard deviation has the same units as the property being measured. The square of standard deviation is called variance (s2). Relative standard deviation is the standard deviation as a fraction of the mean, i.e. S/x. It is sometimes multiplied by 100 and expressed as a percent relative standard deviation. It becomes a more reliable expression of precision.
% Relative Standard Deviation (RSD) = S*100/x
2. Reproducibility
From the reproducibility standard deviation oR or sR it is useful to calculate the ‘Reproducibility limit ‘R’, ‘which enables the analyst to decide whether the difference between duplicate analyses of a sample, determined under reproducibility conditions, is Significant. These calculations can be performed directly with the built-in statistics function of the instrument, if available, or by using a pocket calculator or a PC (Personal Computer) with a suitable software package[51] (e.g. spreadsheet program).
3. Accuracy and recovery
Accuracy can be assessed by analyzing a sample with known concentrations (e.g., a control sample or certified reference material) and Comparing the measured value with the true value as supplied with the material. If certified Reference materials or control samples are not available, a blank sample matrix of interest can be spiked with a known concentration by weight or volume. Standard deviation of slope (Sb) Standard deviation of intercept, (Sa) Intercept values of least squares fits of data are often to evaluate additive errors between or Among different methods. Correlation coefficient, (r) the correlation coefficient r (x, y) is more useful to express the relationship of the chosen Scales. To obtain a correlation coefficient the covariance is divided by the product of the Standard deviation of x and y.[52]
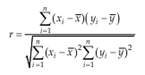
It is best established by comparing the responses of extracted samples at low, medium and high concentrations in replicates at least 6 with those non- extracted standards, which represent 100% recovery.

The correlation coefficient, y-intercept, slope of the regression line, and residual sum of squares should be submitted. A plot of the data should be included. According to the Beers Lambert Law, Absorbance is the ratio of logarithm of Intensity of incident light and Intensity of transmitted light, or A = εCT. The absorbance (A) is proportional to the concentration (C) of the absorbing species, if absorptive (ε) and thickness of the medium (t) are constant. When concentration is in moles per liter, the constant is called molar absorptive. Beers Law limits and Emax values are expressed as µg/ml and moles/cm respectively. Sand ell’s Sensitivity refers to the number of µg of the drug to be determining, converted to the colored product, which in a column solution of cross section 1cmshows an absorbance of 0.001(expressed as µg/cm).[53]
4. Stability
System stability should be determined by replicate analysis of the sample Solution. SIf, on plotting the assay results as a function of time, the value is higher; the maximum duration of the usability of the sample solution can be calculated.
5.Range
The range of an analytical method is the interval between the upper and lower levels (including these levels) that have been demonstrated to be determined with precision, accuracy and linearity using the method as written. The range is normally expressed in the same units as the test results (e.g., percentage, parts per million) obtained by the analytical method. For assay tests, the ICH (5) requires the minimum specified range to be 80 to 120 percent of the test concentration, and for the determination of an impurity, the range to extend from the limit of quantization, or from 50 percent of the specification of each impurity, whichever is greater, to 120 percent of the specification
6. Limit of detection
The limit of detection is the point at which a measured value is larger than the uncertainty associated with it. The limit of detection is frequently confused with the sensitivity of the method. The sensitivity of an analytical method is the capability of the method to discriminate small differences in concentration or mass of the test analyze.
(a) Visual inspection
The detection limit is determined by the analysis of samples with Known concentrations of analyze and by establishing the minimum level at which the Analyze can be reliably detected. [54]
(b) Standard deviation of the response based on the standard deviation of the blank
Measurement of the magnitude of analytical background response is performed by analyzing an appropriate number of blank samples and calculating the standard deviation of these responses.
(c)Standard deviation of the response based on the slope of the calibration curve
A specific calibration curve is studied using samples containing an analyte in the range of the limit of detection. The residual standard deviation of a regression line, or the standard deviation of y-intercepts of regression lines, may be used as the standard deviation.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
7. Limit of quantification:
The limit of quantization is the minimum injected amount that produces quantitative Measurements in the target matrix with acceptable precision in chromatography, typically requiring peak heights 10 to 20 times higher than the baseline noise. If the required precision of the method at the limit of quantization has been specified, the EURACHEM (22) approach Can be used. It is important to use not only pure standards for this test but also spiked matrices that closely represent the unknown samples. For the limit of detection, the ICH (5) recommends, in addition to the procedures as described above, the visual inspection and the standard deviation of the response and the slope of the calibration curve. Figure 3 illustrates the limit of quantization (along with the limit of detection, range and linearity). Figure 4 illustrates both the limit of detection and the limit of quantization.

Figure No 2 Shows Limit of Quantification&Detection
8. Repeatability
From the repeatability standard deviation are it is useful to calculate the ‘repeatability limit ‘r’’, which enables the analyst to decide whether the difference between duplicate analyses of a sample, determined under repeatability conditions, is significant.
9. Measurement uncertainty
Measurement uncertainty is a single parameter (usually a standard deviation with a Coverage factor or confidence interval) expressing the range of values possible on the basis of the measurement result. A measurement uncertainty estimate takes account of all Recognized effects operating on the result; the uncertainties associated with each effect are combined according to well-established procedures. An uncertainty estimate for analytical Chemistry is often termed an ‘uncertainty budget’ and should take into account .The overall, long-term precision of the method;[ 55-56]
· Bias and its uncertainty, including the statistical uncertainty involved in the bias measurements, and the reference material or method uncertainty. It may be necessary to increase the estimate where a significant bias is detected but left uncorrected.
· Calibration uncertainties. As most equipment calibration uncertainties will be Negligibly small by comparison with overall precision and uncertainty in the bias; this needs only to be verified;
· Any significant effects operating in addition to the above. For example, temperature or time ranges permitted by the method may not be fully exercised in validation studies, and their effect may need to be added. Such effects can be usefully quantified by robustness studies (see ‘Ruggedness’ below) or related studies which establish the size of a given effect on the result. Where the contribution of individual effects is important, for example in calibration laboratories.
10. Sensitivity
This is effectively the gradient of the response curve, i.e. the change in instrument response, which corresponds; to a change in analyze concentration. Where the response has been established as linear with respect to concentration, i.e. within the linear range of the method, and the intercept of the response curve has been determined, sensitivity is a useful parameter to calculate and use in formulae for quantization. [57]
11. Ruggedness (or robustness)
Ruggedness is normally evaluated during method development, typically by the originating Laboratory, before collaborating with other laboratories and is a measure how well a method stands up to less than perfect implementation. Performance, and may even result in the method not working at all. These stages should be identified, usually as part of method development, and if possible, their influence on method performance evaluated using ‘ruggedness tests’, sometimes also called ‘robustness tests’[56 57]
CONCLUSION
The efficient development and validation of analytical methods are critical elements in the development of pharmaceuticals. Success in these areas can be attributed to several important factors, which, in turn, will contribute to regulatory compliance. Experience is one of these factors both the experience level of the individual scientists and the collective experience level of the development and validation department .Recent development in pharmaceutical and biotechnological field generates a cumulative demand for analytical methods. Rapid and accurate quantification of the substrate and drug product is important in the process development. Improvements in analytical instrumentation leads to development of new techniques like isocratic and gradient RP-HPLC, which evolved as the primary techniques for the analysis of nonvolatile APIs and impurities. These analytical methods are critical elements of pharmaceutical development so it is very important to develop efficient and accurately validated analytical methods to develop safe and effective drugs. [58]
REFERENCES
1. Swarbrick James., and Boylan James., Encyclopedia of pharmaceutical technology, Volume I, Marcel Dekker Inc., New York, (1998), 217 - 224.
2.Beckett,A.H., Stenlake J.B., Practical Pharmaceutical Chemistry, (1997), 4th edition, Part 2, CBS Publishers and distributors, 275-337.
3. Glenn A.L., Journal of pharmacy and pharmacology, (1960), 12,598-608.
4.Jain H.K., Agrawal R.K., Indian Journal of pharmaceutical sciences, (2002), 64(1), 88-71.
5.Shankar M.B., Mehta F.A., Bhatt K.K., Mehta R.S., and Geetha M., Indian Journal of pharmaceutical sciences, 65(2), 167-170.
6.Skoog DA, West DM, Holler FJ (1996) Fundamentals of analytical chemistry. (8thEdn), Fort Worth: Saunders College Pub.
7.Song HH, Choi KS, Kim CW, Kwon YE (2009) Pharmacokinetic Profiles of Two Branded Formulations of Piroxicam 20mg in Healthy Korean Volunteers by a Rapid Isocratic HPLC Method. J Bioequiv Availab 1: 074-079.
8. Nanjwade BK, Ali MS, Nanjwade VK, Manvi FV (2010) Effect of Compression Pressure on Dissolution and Solid State Characterization of Cefuroxime Axetil. J Anal Bioanal Techniques 1:112.
9. Yue PF, Yuan HL, Yang M, Zhu WF (2009) Preparation, Char-acterization and Pharmacokinetics in Vivo of Oxymatrine-Phospholipid Complex. J Bioequiv Availab 1: 099-102.
10. Maithani M, Singh R (2011) Development and Validation of a Stability-Indicating HPLC Method for the Simultaneous Determination of Salbutamol Sulphate and Theophylline in Pharmaceutical Dosage Forms. J Anal Bioanal Techniques 1:116.
11. Chitlange SS, Chaturvedi KK, Wankhede SB (2011) Development and Validation of Spectrophotometric and HPLC Method for the Simultaneous Estimation of Salbutamol Sulphate and Prednisolone in Tablet Dosage Form. J Anal Bioanal Techniques 2:117.
12. Saber AL, Amin AS (2011) Utility of Ion-Pair and Charge Transfer Complexation for Spectrophotometric Determination of Domperidone and Doxycycline in Bulk and Pharmaceutical Formulations. J Anal Bioanal Techniques 1:113.
13.Bai L, Ma Z, Yang G, Yang J, Cheng J (2011) A Simple HPLC Method for the Separation of Colistimethate Sodium and Colistin Sulphate. J Chromatograph Separat Techniq 1:105.
14.Babu ARS, Thippeswamy B, Vinod AB (2011) Determination of Tacrolimus in Rat Whole Blood Utilizing Triple Quadrupole LC/MS. J Anal Bioanal Techniques 2:118.
15.Junior EA, Duarte LF, Pereira R, Pozzebon JM, Tosetti D, et al. (2011) Gabapentin Bioequivalence Study: Quantification by Liquid Chromatography Coupled to Mass Spectrometry. J Bioequiv Availab 3: 187-190.
16.Hsieh CL, Wang HE, Ker YB, Peng CC, Chen KC, et al. (2011) GC/MS Determination of N-butyl-N-(3-carboxypropyl) Nitrosamine (BCPN) in Bladder Cancers - The Skewed Molecular Interaction Caused Retention Time Shift. J Anal Bioanal Techniques 1:115.
17.Ekeberg D, Norli HR, Stene C, Devle H, Bergaust L (2010) Identification of Brominated Flame Retardants in Sediment and Soil by Cyclohexane Extraction and Gas Chromatography Mass Spectrometry. J Chromatograph Separat Techniq1:102
18.Ravali R, Phaneendra M, Bhanu Jyothi K, Ramya Santhoshi L, Sushma K (2011) Recent Trends in Analytical Techniques for the Development of Pharmaceutical Drugs. J Bioanal Biomed R1: 002.
19. M.S. Tswett, TR Protok, Otd. Biol., 14 (1903, publ. 1905) 20.
20.Verzele M, Dewaele C (1985) Preparative High Performance Liquid Chromatography, A Practical Guidline. TEC Dent, Belgium.
21.Dhaneshwar SR, Salunkhe JV, Bhusari VK (2010) Validated HPTLC Method for Simultaneous Estimation of Metformin Hydrochloride, Atorvastatin and Glimepiride in Bulk Drug and Formulation. J Anal Bioanal Techniques 1:109.
22.Abdelkawy M, Metwaly F, El Raghy N, Hegazy M, Fayek N (2011) Simultaneous determination of Ambroxol Hydrochloride and Guaifenesin by HPLC, TLCSpectro densitometric and multivariate calibration methods in pure form and in Cough Cold Formulations. J Chromatograph Separat Techniq 2:112.
23. Bari SB, Bakhshi AR, Jain PS, Surana SJ (2011) Development and Validation of Stability-Indicating Tlc Densitometric Determination of Ropinirole Hydrochloride in Bulk and Pharmaceutical Dosage Form. Pharm Anal Acta 2:125.
24.Jain PS, Khatal RN, Jivani HN, Surana SJ (2011) Development and Validation of TLC-densitometry Method for Simultaneous Estimation of Brimonidine tartrate and Timolol maleate in Bulk and Pharmaceutical Dosage Form. J Chromatograph Separat Techniq 2:113.
25.Skoog DA, Leary JJ (1992) Principles of Instrumental Analysis. Harcourt Brace College Publishers.
26.Subbaiah PR, Kumudhavalli MV, Saravanan C, Kumar M, Chandira RM (2010) Method Development and Validation for estimation of Moxifloxacin HCl in tablet dosage form by RP-HPLC method. Pharm Anal Acta 1:109.
27. Sultana N, Arayne MS, Naveed S (2011) RP-HPLC Method for Simultaneous Determination of Captopril and Diuretics: Application in Pharmaceutical Dosage Forms and Human Serum. J Chromatograph Separat Techniq 2:109.
28. Krstulovic AM, Brown PR (1982) Reversed-Phase High Performance Liquid Chromatography, Wiley, New York.
29.Sawant L, Prabhakar B, Pandita N (2010) Quantitative HPLC Analysis of Ascorbic Acid and Gallic Acid in Phyllanthus Emblica. J Anal Bioanal Techniques 1:111.
30.El-Sayed AAY, Mohamed KM, Hilal MA, Mohamed SA, Aboul-Hagag KE, et al. (2011) Development and Validation of High-Performance Liquid Chromatography-Diode Array Detector Method for the Determination of Tramadol in Human Saliva. J Chromatograph Separat Techniq 2:114.
31.Reddy YR, Kumar KK, Reddy MRP, Mukkanti K (2011) Rapid Simultaneous Determination of Sumatriptan Succinate and Naproxen Sodium in Combined Tablets by Validated Ultra Performance Liquid Chromatographic Method. J Anal Bioanal Techniques 2:121.
32.Naveen Kumar Reddy G, Rajendra Prasad VVS, Maiti NJ, Nayak D, Prashant Kumar M (2011) Development and Validation of a Stability Indicating UPLC Method for Determination of Moxifloxacin Hydrochloride in Pharmaceutical Formulations. Pharm Anal Acta 2:142.
33.Nanjwade BK, Patel DJ, Parikh KA, Nanjwade VK, Manvi FV (2011) Development and Characterization of Solid-Lipid Microparticles of Highly Insoluble Drug Sirolimus. J Bioequiv Availab 3: 011-015.
34. Yi SJ, Shin HS, Yoon SH, Yu KS, Jang IJ, et al. (2011) Quantification of Ticlopidine in Human Plasma Using Protein Precipitation and Liquid Chromatography Coupled with Tandem Mass Spectrometry. J Bioanal Biomed 3: 059-063.
35. Rajender G, Narayana NGB (2010) Liquid Chromatography-Tandem Mass Spectrometry Method for Determination of Paclitaxel in Human Plasma. Pharm Anal Acta 1:101.
36.Yang G, Liu Y, Liu H, Yang C, Bai L, et al. (2010) Preparation of a Novel Emulsion-Templated MIP Monolith and its Application for on Line Assay of Nifedipine in Human Plasma. J Chromatograph Separat Techniq 1:103.
37.Moreno RA, Sverdloff CE, Oliveira RA, Oliveira SE, Borges DC, et al. (2009) Comparative bioavailability and pharmacodynamic aspects of cyclobenzaprine and caffeine in healthy subjects and the effect on drowsiness intensity. J Bioequiv Availab 1: 086-092.
38. Puri A, Mehdi B, Panda NB, Dhawan GDPS (2011) Estimation of Pharmacokinetics of Propofol in Indian Pateints by HPLC Method. J Anal Bioanal Techniques 2:120.
39.Remsberg CM, Yáñez JA, Vega-Villa KR, Davies NM, Andrews PK, et al. (2010) HPLC-UV Analysis of Phloretin in Biological Fluids and Application to Pre- Clinical Pharmacokinetic Studies. J Chromatograph Separat Techniq 1:101.
40.Sarker SD, Lafont R, Girault JP, Sik V, Dinan L (1998) Pharmaceutical Biology. 36: 202-206.
41. Sievert P, Hancock WS (1996) new methods in peptide mapping for the characterization of proteins, CRC press, Inc., New York.
42.Furusawa N (2001) Determining the procedure for routine residue monitoring of sulfamethazine in edible animal tissues. Biomed Chromatogr 15: 235-239.
43.Guiochon G, Katti A (1987) Preparative Liquid Chromatography. Chromatographia 24: 165.
44. Bao Y, Li C, Shen H, Nan F (2004) Determination of saikosaponin derivatives in Radix bupleuri and in pharmaceuticals of the Chinese multiherb remedy xiaochaihu-tang using liquid chromatographic tandem mass spectrometry. Anal Chem 76: 4208-4216.
45.Liu L, Cheng Y, Zhang H (2004) Phytochemical analysis of anti-atherogenic constituents of Xue-Fu-Zhu-Yu-Tang using HPLC-DAD-ESI-MS. Chem Pharm Bull 52: 1295-1301.
46.Liu SJ, Liu ZX, Jug WZ, Zhou L, Chen M, et al. (2010) Development and Validation of a Liquid Chromatographic/ Mass Spectrometric Method for the Determination of Saikosaponin a in Rat Plasma and its Application to Pharmacokinetic Study. J Anal Bioanal Techniques 1:104.
47. Breda C.A., Breda M., Frigerio E., Bioanalytical method validation: a risk-based approach?, Journal of Pharmaceutical and Biomedical Analysis,35, 2004, 887–89
48.Nakashima Kenichiro, High-Performance Liquid Chromatography of drug of abuse in biological samples, Journal of Health Science, 51(3) 272-277 (2005)
49.Boulanger B., Chiap P. Dewe W., Crommen J., Hubert Ph., An analysis of the SFSTP guide on validation of chromatographic bioanalytical methods: progresses and limitations Journal of Pharmaceutical and Biomedical Analysis,32, 2003,753-765
50. Causon Roger, Validation of chromatographic methods in biomedical analysis viewpoint and discussion, Journal of Chromatography B, 689 (1997) 175-180
51.Hartmann C., Smeyers-Verbeke J., Massart D. L., McDowall R.D., Validation of bioanalytical chromatographic methods, Journal of Pharmaceutical and Biomedical Analysis17, 1998,193–218
52. G. C. Hokanson, A life cycle approach to the validation of analytical methods during pharmaceutical product development, Part II: Changes and the need for additional Validation, Pharm.Tech. Oct. 1994, pp. 92–100.
53. J. M. Green, A practical guide to analytical method validation, Anal. Chem. News & Features, 1 May 1996, pp. 305A–309A.
54. B. Renger, H. Jehle, M. Fischer and W. Funk, Validation of analytical procedures in pharmaceutical analytical chemistry: HPTLC assay of theophylline in an effervescent tablet, J. Planar Chrom. 8:269–278 (July/August 1995).
55. Wegscheider, Validation of analytical methods, in: Accreditation and quality assurance In analytical chemistry, edited by H. Guenzler, Springer Verlag, Berlin (1996).
56. S. Seno, S. Ohtake and H. Kohno, Analytical validation in practice at a quality control laboratory in the Japanese pharmaceutical industry, Accred. Qual. Assures. 2:140–145 (1997).
57. AOAC Peer-Verified Methods Program, Manual on policies and procedures, Arlington,
58. A report by Jay Breaux , Kevin Jones and Pierre Boulas AAI Development ServicesAnalytical Method Development and Validation
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






