

 About Authors:
About Authors:
Kapil Sharma*, Priyanka Sharma**
*M.Pharm, Yaresun Pharmaceutical Pvt Ltd, India
**M.sc, Yaresun Pharmaceutical Pvt Ltd,
Rajasthan, India.
Aim: - To search Pharmaceutical medicine (New, Safe & Effective) to enhance health and benefits of patients & community.
Trends:-Usually search a pharmaceutical medicine is starting from “Drug Discovery and Drug Development” (D4) or Pharmaceutical R&D.
D4 is very long, very costly and very complex process, so mostly companies put together D4 plan. Drug Discovery and Drug Development is the treds of searching a Pharmaceutical medicine
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1259
Generation of R & D:- Now this is the time of fourth generation of Pharmaceutical R & D.
I-Generation –Early 1990s
II- Generation –Early 1995s
III- Generation –Early 2003s
IV- Generation –Present time
Cost:-Total cost of D4 is more than US$850 million.
Emphasis:- Total emphasis of D4 is searching a Pharmaceutical medicine.
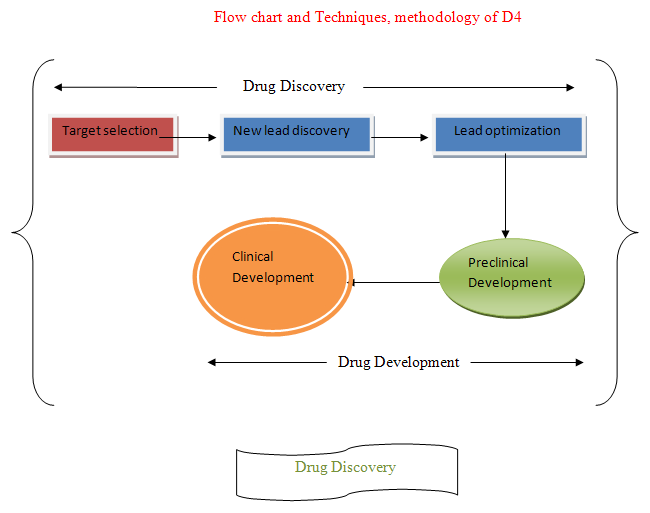
Drug Discovery and Drug Development:- Made from two world
Drug Discovery + Drug Development
Drug Discovery:-Involve
Target selection + New lead discovery + Lead optimization
Drug Development:- Involve
Preclinical development(Animal testing) + Clinical development (Human testing)/Clinical trials
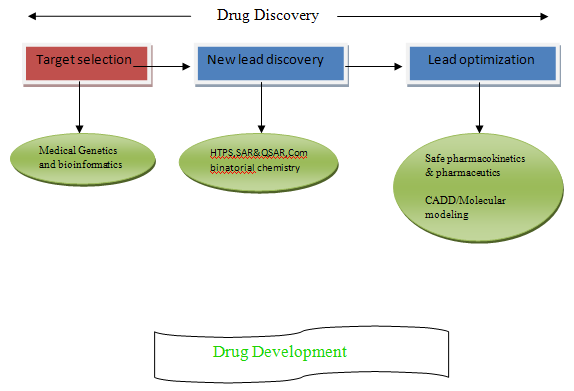
Target selection:-Involves choosing a disease to treat & then developing a model for that disease.
It helps to find the right drugs for the right patients.
It uses application of medical genomics and bioinformatics
New lead discovery:-Involves developing of assay & screening of large compounds and Collections.
It helps determination of lead structure.
It uses different techniques like-
HTP Screening (High through put screening)
SAR & QSAR (Structure activity relationship/Q-Quantitative SAR)
Combinatorial chemistry & Bio-synthetic chemistry
Lead Optimization:-Involves modification in the determined lead structure by safe
Pharmacokinetic and pharmaceutics study.
Helps to improve activity to reduce side effects and to improve performance
It uses CADD(Computer aided drug design)/Molecular modeling.
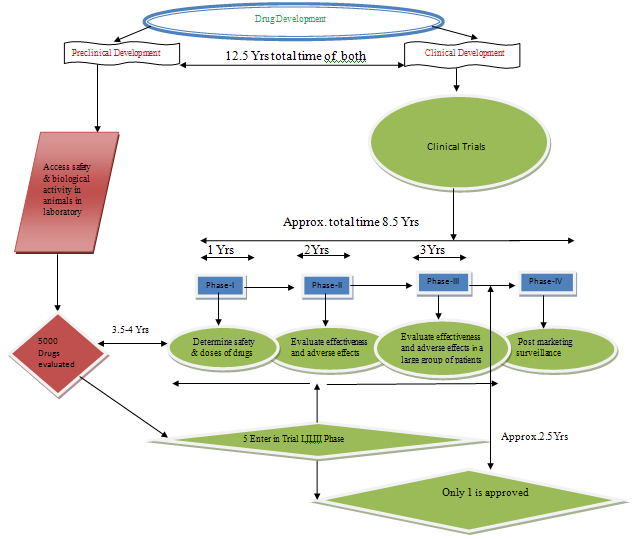
Preclinical development:-Access safety & biological activity in animals in laboratory.
Involves- Formulation (For suitable dosage forms of drugs)
Animal Pharmacology
Animal Pharmacokinetics
Animal Toxicology
Clinical development:-Involves four PhasesI to IV as termed “Clinical Trials”
Phase-I:-Determine safety & doses of drugs
Taking 20-80 healthy volunteers
Getting information about Pharmacokinetics and Pharmacology
Side-effects with increasing doses
Only 70% of experimental drugs passes phase-I trial
Phase-II:-Evaluate effectiveness and adverse effects in mild, moderate and sever cases of the diseases
Taking 100 to 300 patients
Drug effectiveness in all expected indications
Only 35% of experimental drugs passes phase-II trial
Phase-III:-Verify the drugs’effectivness & adverse effect in a large group of patients over a
Longer period of exposure to establish safety and efficacy
Taking several hundred to several thousand patients (involves subgroups likes
Children, elderly, liver & kidney impairments functions patients)
Taking decisions about marketing of drug is allowed.
Phase-IV:-Post marketing surveillance
Identify any light adverse effects after marketing.
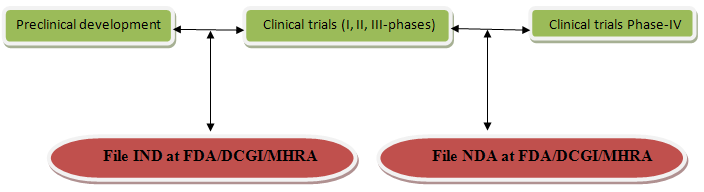
Files for INDA:- After completing preclinical development the company files an IND/INDA With FDA/DCGI/MHRAto begin testing the drug in human. The IND become effective if FDA/DCGI/MHRA does not disapprove it within 30 days.
Files for NDA:- For marketing, approval is taking from appropriate regulatory authorities. After completing of phase-III of clinical trial company analyze data and file on NDA to FDA/DCGI/MHRA. The regulatory authority (FDA/DCGI/MHRA) grants a product licence and after that company can be marketed.
IND/INDA-Investigational new drug application
FDA-Food & drug administration (USA)
MHRA-Medicines & healthcare products regulatory agency(UK) advised by CSM (Committee on safety of medicines)
DCGI-Drug controller general of India (INDIA) under CDSCO (Central drugs control Organization)
NDA:-New drug application
Generic drug product:-Generally a patent for a new drug is 20 Yrs.
Once the patent is expired other company/manufacturer can produce the
Product (Called a generic version) and sell it at a lower cost.
Generic use the same active ingredients and work the same in the body.
Generic drug products are less expensive because generic drug
Manufactures don’t have the investment costs of the D4.
ANDA:-Abbreviated New Drug Application
Usually ANDA is submitted to regulatory bodies to obtain the approval to market a Generic drug product.
Generic drug application are termed ‘’abbreviated.’’
Generic drug company/manufacturer focuses on bioequivalence data, chemistry, microbiological data, plant inspection and drug labeling information.
[adsense:468x15:2204050025]
Major segments-
Anti-infective is estimatedto be the major therapeuticsegment accounting for14.7% of the total pharmamarket in India.The major sub-segments within this category are Cephalosporin, Quinolones, Penicillin and Macrolides. Cardiovascular drugs are the next major therapeutic segment contributing 11.1% of the total sales.
Present Problems in D4
*Indian Pharma companies are on a global Trends, specially gaining importance in the generics market policies.
* Indian companies de-risk their R&D by cut off their R&D unit into a separate portions.
*Mostly drugs are under price control.
* Mostly Drugs in some state are free of cost as govt. supply.
Industry Trend
• The strengthening of Indian Pharma companies to:
1. Protection by Launching patents of drugs in India
2. Investment to increasing R&D presence in India
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
PREFORMULATION: A NEED FOR DOSAGE FORM DESIGN
The Concept of Preformulation:-
Almost all drugs are marketed as tablets, capsules or both. Prior to the development of these major dosage forms, it is essential that pertain fundamental physical and chemical properties of the drug molecule and other divided properties of the drug powder are determined. This information decides many of the subsequent events and approaches in formation development. This first learning phase is known as preformulation.
Definition:-
Preformulation involves the application of biopharmaceutical principles to the physicochemical parameters of drug substance are characterized with the goal of designing optimum drug delivery system.
Before beginning the formal preformulation programs the preformulation scientist must consider the following factors:-
- The amount of drug available.
- The physicochemical properties of the drug already known.
- Therapeutic category and anticipated dose of compound.
- The nature of information, a formulation should have or would like to have.
Preformulation drug characterization in a structured program:-
|
Test |
Method/ function Characterization |
|
FUNDAMENTAL |
|
|
1) UV spectroscopy |
Simple assay |
|
2) Solubility |
Phase solubility/ purity |
|
a) Aqueous |
Intrinsic & pH effect |
|
b) pKa |
solubility control , salt formation |
|
c) Salt |
Solubility, hygroscopicity & stability |
|
d)Solvents |
Vehicles & Extraction |
|
e) ko/ w |
Lipophillicity, structure activity |
|
f) Dissolution |
Biopharmacy |
|
3) Melting point |
DSC-polymorphism hydrate & solvent |
|
4) Assay development |
UV, HPLC, TLC |
|
5) Stability |
|
|
In Solution |
Thermal, hydrolysis, pH |
|
In solid state |
Oxidation, proteolysis metal ion |
|
Derived |
|
|
6) Microscopy |
Particle size and morphology |
|
7) Bulk density |
Tablet and capsule formation |
|
8) Flow properties |
Tablet and capsule formation |
|
9) Compression properties |
Acid / excipient choice |
|
10) Excipient compatibility |
Preliminary screen by DSC, Conformation by TLC |
UV Spectroscopy :-
The first requirement of any preformulation study is the development of a simple analytical method for quantitative estimation in subsequent steps. Most of drugs have aromatic rings and/or double bonds as part of their structure and absorb light in UV range, UV spectroscopy being a fairly accurate and simple method is a performed estimation technique at early preformulation stages. The absorption Co-efficient of the drug can be determined by the formula:-
E = AF / X
Where , A = Asborbance
F= dilution factor
X = weight of drug (mg)
It is now possible to determine concentration of drug in any solution by measuring absorbance.
C = AF / E mg/ ml
Characterization of drug molecules is very important step at the preformulation phase of product development. Following studies are conducted as basic preformulation studies, special studies are conducted depending on the type of dosage form and the type of drug molecules.
1) Solubility determination
2) pKa determination
3) Partition co-efficient
4) Crystal properties and polymorphism
5) Practical size, shape and surface area.
6) Chemical stability profile.
Solubility Determination:-
The solubility of drug is an important physicochemical property because it affects the bioavailabilty of the drug, the rate of drug resale into dissolution medium and consequently, the therapeutic efficiency of the pharmaceutical product.
The solubility of the molecules in various solvents is determined as a first step. This information is valuable is developing a formulation. Solubility is usually determined in variety of commonly used solvents and some oils if the molecule is lipophillic.
The solubility of material is usually determined by the equilibrium solubility method, which employs a saturated solution of the material, obtained by stirring an excess of material in the solvent for a prolonged until equilibrium achieved :-
Common solvents used for solubility determination are:-
·Water, ·Polyethylene Glycols, ·Propylene Glycol, ·Glycerin, ·Sorbitol, ·Ethyl Alcohol, ·Methanol, ·Benzyl Alcohol, ·Isopropyl Alcohol, ·Tweens, ·Polysorbates, ·Castor Oil, ·Peanut Oil, ·Sesame Oil, ·Buffer at various pHs
Aqueous Solubility:-
The availability of a drag is always limited and the preformulation scientist may only have 50 mg. Solubility dictates the ease with which formulation for oral gavages and intravenous injection studies in animals are obtained the pKa allives the informed of pH to maintain solubility and to choose salts required to achieve good bioavailability from the solid state and improve stability and powder properties.
Intensic Solubility (Co):-
An increase in solubility in acid compared to aqueous solubility suggests a weak base and an increase in alkali, a weak acid. An increase in acidic and alkaline solubility suggests either impotence or zuitter ion behavior. In this case there will be two pKa’s, one acidic & one basic. When the purity of the drug sample can be assured the solubility obtained in acid for a weak acid or alkali for a weak base can be assured to be the intrinsic solubility (Co.) i.e. the fundamental solubility when completely unionized. The solubility should ideally be measured at two temperatures.
1)4C to ensure physical stability and entered short term storage and chemical stability unit more definitive data are available. The minimum density of water occurs at 4C. This leads to a minimum aqueous solubility.
2)37C to support biopharmaceutral evaluation.
pKa Determination:-
Determination of the dissociation content for a drug capable of ionization within a ph rang of 1 to 10 is important since solubility and consequently absorption, cab be altered by orders of magnitude with changing pH. The Henderson – Hasseslebach equation provides an estimate of the ionized and un ionized durg concentration at a particular pH.
For acidic compounds
pH = pKa + log (un-ionized drug]) / [ionized drug])
Partition Coefficient :-
Partition Coefficient (oil/ water) is a measure of a drug’s lipophilicity and an indication of its ability to cross cell membranes. It is defined as the ratio of unionized drug distributed between the organic and aqueous phases at equilibrium.
P o/w = (C oil / C water) equilibrium.
For series of compounds, the partition coefficient can provide an empiric handle in screening for some biologic properties. For drug delivery, the lipophilic/ hydrophilic balance has been shown to be a contributing factor for the rate and extent of drug absorption. Although partition coefficient data alone does not provide understanding of in vivo absorption, it does provide a means of characterizing the lipophilic/ hydrophilic nature of the drug.
Since biological membranes are lipoidal in nature. The rate of drug transfer for passively absorbed drugs is directly related to the lipophilicity of the molecule. The partition coefficient is commonly determined using an oil phase of octanol or chloroform and water.
Drugs having values if P much greater than 1 are classified as lipophilic, whereas those with partition coefficient much less than 1 are indicative of a hydrophilic drug.
Although it appears that the partition coefficient may be the best predictor of absorption rate, the effect id dissolution rate, pKa and solubility on absorption must not be neglected.
Dissolution :-
The dissolution rate of the a drug is only important where it is the rate limiting step in the absorption process. Kaplan suggested that provided the solubility of a drug exceded to mg/ ml at pH , 7 no bioavailability or distinction related problems were to be expected. Below / mg/ ml such problems were quite possible and salt formation could improve absorption and solubility by controlling the pH of the microenvironment, independently of the drug and dosage forms position within the GI ireat.
Intrinsic Dissolution Rate :-
When dissolution is controlled solely by diffusion the rate of diffusion is directly proportional to the saturated concentration of the drug in solution under these conditions the rate constant K1 is defined by
K1 = 0.62 D2/3 v 1/6 w1/2
Where, V is the kinemative viscosity
W is the anguter velocity of a rotating disc of drug.
Common Ion Effect :-
A common ion significantly reduces, the solubility of a slightly soluble electrolyte. The ‘selling out’ results from the removal of water molecules as solvent owing to the completing hydration of other ions. The reverse process ‘salting in’ qries with large anions e.g. benzoate, salivate which open the water structure. These hydro topics increase the solubility of properly water soluble compounds such as diazepam.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Melting Point :-
The melting point of a drug can be measured using three techniques :-
1)Capillary Melting
2)Hot Stage Microcopy
3)Differential scanning calorinetry or thermal Anaylysis.
Capillary Melting :-
Capillary melting gives information about the melting range but it is different to assign an accurate melting point.
Hot Stage Microcopy:-
This the issued observation of melting under a microscope equipped with a heated and lagged sample stage. The heating rate is controllable and upto three transitions can e registered.
Differential Scanning Calorimeltry and thermal analysis :-
Differential thermal analysis (DTA) measures the temperature difference between the sample and a reference as a function of temperature or time when heating at a constant rate differential scanning calorinetry (DSC) is similar to DTA except that the instrument measures the amount of energy required to keep the sample at the same temperature as the reference i.e. it measures the enthalpy of transition.
Crystal Properties and Polymorphism :-
Many drug substance can exit in more than one crystalline from with different space lattice arrangements. This property is known as polymorphism. Polymorphs generally have diffrent melting points, x-ray diffraction patterns and solubility even though they are chemically identical.
Differences in the dissolution rates and solubilities of different polymorphic forms of a given drug are very commonly observed. When the absorption of a drug is dissolution rate limited, a more soluble and faster-dissolving from may be utilized to improve the rate and extent of bioavailability.
For drugs pane to degradation in the solid state, physical form of the drug influences degradation. Selection of a polymorph that is chemically more stable is a solution in many cases. Different polymorph also lead to different morphology, tensile strength and density of power bed which all contribute of compression characteristics of materials. Some investigation of polymorphism and crystal habit of a drug substance as it relates to pharmaceutical processing is desirable during its Preformulation evaluation especially when the active ingredient is expected to constitute the bulk of the tablet mass. Although a drug substance may exist in two or more polymorphic forms, only one form is theromdynamically stable at a given temperature and pressure. The other forms would convert to the stable form with time. In general, the stable polymorph exhibits the highest melting point , the lowest solubility, and the maximum chemical stability. Various techniques are available for the investigation of the solid state. These include microscopy (including hot stage microcopy), infrared spectrophotometry, single-crystal x-ray and x-ray power diffraction, thermal analysis, and dilalometry.
Particle Size, Shape and Surface Area:-
Bulk flow, formulation homogeneity, and surface-area controlled processes such as dissolution and Surface morphology of the drug particles. In general, each new drug candidate should be tested during Preformulation with the smallest particle size as is practical to facilitate preparation of homogeneous samples and maximize the drug’ s surface area for interactions.
Various chemical and physical properties of drug substances are affected by their particle size distribution and shapes. The effect is not only on the physical properties of solid drugs but also, in some instances, on their biopharmaceutical behavior. It is generally recognized that poorly soluble drugs showing a dissolution- rate limiting step in the absorption process will be more readily bio available when administered in a finely subdivided state rather than as a coarse material.
In case of tablets, size and shape influence the flow and the mixing efficiency of powders and granules. Size can also be a factor in stability: fine materials are relatively more open to attack from atmospheric oxygen, the humidity, and interacting excipients than are coarse materials.
- Determination of particle size
-Determination of surface area
Particle size Determination:-
Though microscopy is the simplest technique of estimating size ranges and shapes, it is to slow for quantitative determination the material is best observed as a suspension in non dissolving fluid. Saving is less useful technique at preformulation storage due to lack of bulk material. Andreason pipette is based on the rate difference of sedimentation of different particles, but techniques like this are seldom used due to their tedious nature instruments based on light scattering, (Royco), light blockage (HIAC) and blockage of electrical conductivity path (coulter counter) are available.
Surface Area Determination:-
Surface area is most commonly determined based on brunaver emette teller (BET) theory of adsorption. Most substances adsorb a mono molecular layer of gas under certain conditions of partial pressure of gas and temperature. Knowing the monolayer capacity of adsorbent and the area of absorbale molecule, the surface area can be calculated the adsorption process is carried out with nitrogen at-195 degree Celsius at a partial pressure attainable when nitrogen is in a 30% temperature with an inert gas (helium). The adsorption takes place by virtue of vander wall’s forces.
Power Flow Properties:-
When limited amounts of drugs are available Power flow properties can be evaluated by measurements of bulk density and angle of repose. Changes in particles size, and shape are generally very important an increase in crystal size or a more uniform shape will lead to a small angle or rpose and a smaller Carr’s index.
Bulk Density :
Knowledge of absolute and bulk density of the drug substance is Very useful in Having some idea as to the size of final dosage form the density of solids also of affects their flow Properties Carr’s compressibility index can be used to predict the flow properties based on density measurement.
Carr’s index (%) = Tapped density – Pored density *100
Tapped density
A similar index has been defined by Hausner :
Hausner ratio = Tapped density
Pored density
Angle of repose:-
The maximum angle which is formed b/w the surface of a pile of powder and horizontal surface is called the angle of repose.
Relationship between flow, angle of repose, Carr’s index fee power flow
|
Flow |
Angle of repose |
Carr’s index ( % ) |
|
Excellent |
<25 |
5-15 |
|
Good |
25-30 |
12-16 |
|
Fair to passable |
30-40 |
18-21 |
|
Poor |
> 40 |
23-35 |
|
Very Poor |
33-38 |
|
|
Extremely Poor |
>40 |
|
Chemical stability profile:
Preformulation stability studies are usually the first quantitative assessment of chemical stability of a new drug. These studies include both solution and solid state experiments under condition typical for the handing, formulation, storage, and administration of a drug candidate as well as stability in presence of other recipients.
Factor effecting chemical stability critical in rational dosage form design include temperature, pH and dosage form diluents. The method of sterilization of potential product will be largely dependent on the temperature stability of the drug. Drugs having decreased stability at elevated temperatures cannot be sterilized by autoclaving but must be sterilized by another means, e.g., filtration. The effect of pH on drug stability is important in the development of both oral administration must be protected from the highly acidic environment of the stomach. Buffer selection for potential dosage forms will be largely based on the stability characteristic of the drug.
- Solid state stability
- Solution phase stability
- Compatibility studies : stability in the Presence of excipients
- Typical stability protocol for anew Chemical Entity
Solid state stability:-
Chemical instability normally results from either of the following reaction :- hydrolysis, oxidation, photolysis and pyrolysis, Chemical structure of the drug is the determination of drug to either of these attacks. Esters and lactase and to lesser extent, amides are to prone to solvolysis . Instauration or electron rich centre in the structure make the molecule vulnerable for free radical mediated or photo-catalysed oxidation. physical properties of drugs. Amorphous materials are less stable than their crystalline forms. Denser materials are more stable to ambient stress.
Elevated temperature studies:-
The elevated temperatures commonly used are 40, 50, and 60 degree centigrade with ambient humidity. The samples stored at highest temperature are observed weekly for physical and chemical changes and compared to an appropriate control . If a substantial change is seen, samples stored at lower temperature are examined . If no changesisseen after 30 days at 60 degree centigrade, the stability prognosis is excellent .
Stability under high humidity conditions :-
Solid drug samples can be exposed to different relative humidity conditions by keeping them in laboratory desiccators containing saturated solutions of various salts. The closed desiccators in turn are kept in oven to provide constant temperature. The preformulation data of this nature are useful in determining if the material should be protected and stored in controlled low humidity environment or if non aqueous solvent be used during formulation.
Photolytic stability :-
Many drugs fade or dorpen on exposure light. Though the extent of degradations small and limited to the exposed surface area, it presentsanaesthetic problem. Exposure of drug 400 and 900 foot-candles of illumination for 4 and 2 week periods respectively is adequate to provide some idea of photosensitivity. Resulting data may be useful in determining if an amber colored container is required or if color masking bye should be used in the formulation .
Stability to Oxidation :-
Drug’s sensitivity to oxidation can be examined by exposing it to atmosphere of high oxygen tension. Usually a 40% oxygen atmosphere allows for rapid evaluation. A shallow layer of drug exposed to a sufficient headspace volume ensures that the system is not oxygen limited. Samples are kept in desiccators equipped with three-way stop cocks, which are alternatively evacuated and flooded with desired atmosphere. The process is repeated 3 or 4 times to ensure 100% desired atmosphere. Results may be useful in predicting if an antioxidant is required in the formulation or if the final product should be packaged under inert atmospheric conditions.
Compatibility studies :-
The knowledge of drug excipients interaction is useful for the formulation to select appropriate excipients. The described preformulation screening of drug excipients interaction requires only 5mg of drug in a 50% mixture with the excipients to maximize the likelihood of obscuring an interaction . Mixtures should be examined under nitrogen to ultimate oxidation and paralytic effect at a standard heating rate on DSC, over a temperature range, which will encompass any thermal changes due to both the drug and appearance or disappearance one or more peaks in themogrames of drug excipient mixtures are considered of indication of interaction.
Solution phase stability:
As compared with the dry form, the degradation is much rapid in solution form. It is important ascertain that the drug doesn’t degrade when exposed to GI fluid. The pH based stability study, using different stimulator GI condition can be designed. A poor solution stability of drug may urge the formulator to choose a less soluble salt form, provided the bioavailability is not compromised
Absorption behavior:
It is essential to test the in vivo behavior of the new drug for successful formulation of a dosage from good bioavailability. Partial in vivo and in vitro test are designed to study pharmacokinetic profile of the drug.
CONCLUSION:
Preformulation studies have a significant part to play in anticipating formulation problems and identifying logical path in both liquid and solid dosage form technology. The need for adequate drug solubility cannot be overemphasized. The most appropriate salt for development. Stability studies in solution will indicate the feasibility of parental or other liquid dosage form and can identify methods of stabilization. In parallel solid-state stability by DSC, TLC and HPLC in the presence of tablet and capsule excipient will indicate the most acceptable vehicles for solid dosage form.
By comparing the physicochemical properties of each drug candidate with in a therapeutic group, the preformulation scientist can assist the synthetic chemist to identify the optimum molecule, provide the biologist with suitable vehicles to elicit pharmacological response and advise the bulk chemist about the selection and production of the best salt.
GENERAL INSTRUMENTAL REQUIREMENTS OF D4:-
For research methods and labs-
Double beam uv-visiblespectroscopy
Ir- spectroscopy
NMR (1Hnmr, 13Cnmr) spectroscopy
Mass- spectroscopy
Flourimetry
Electrophoresis
Chromatography (liquid chromatography/hplc, GC, hptlc, TLC, paper chromatography, column chromatography)
X-rays crystallography, muffle furnace, refractometry, uv-chamber, potentiometer
Polarimeter
Lab oven
Hot plate
Agitator
Rotatory shaker
Dtapparatus
Dissolution test apparatus
Ph-meter
Sonicator
Vaccum pump
Filtering assembly
Distillation assembly
Hardness-tester
Varnier caliper
Karlfischer auto titrator
Bulk density apparatus
Micrometer/screw gauge
MP & BP apparatus
Hot plate with magnetic stirrer
Friability apparatus
Ir-moisture balance,
0.1mg electronic balance
Colorimeter
Flame photometer
Microbiological labs setup
Stability testing labs
Animal Testing-Depends upon models and drug selections
For formulations-
Tablet-rapid mixer granulator, fluid bed drier, tablet punching machine 16/27/35 stations,multimill,stiffer s.s.s,stifer sieve 30 inch 12/16/20/40/60,multimill sieve size 10mm/3mm/2mm/1mm/15mm,octagonal blender ,coating psn,colloid mill, tablet dedusting machine, dies & punches, dust extracting unit,dehumidifier,starch paste castele,pressure vessel with gauge with hydrodynamic atomizing spray gun with stand & clamp, polishing pan for coating pan, tablet inspection tablet c belt and packing machine(blister/strip/alu-alu packs)
Capsule & dry syrup-s.s. Sifter, double cone blender(different capacity),semi/-automatic cap fill machine, capsule polishing and inspection machine, tray drier 24/48,aunger filling machine,p.p.cap sealing machine, labeling/printing machine,dehumidifier,capsules filling machine, capsule loader with change plates for0,1,2
Liquid oral section- sugar preparation tank .s.s jacket, tank s.s.1000/500/300/100/50lts,filter press with pump, automatic liquid bottle washing, filling/sealing/printing /gumming machine, s.s.stirer with stand, s.s.emulsifier with stand, S.S. Transfer gear pump
References:
1. H. Brittain, Physical Characterization of Pharmaceutical Solids, Marcel Dekker, Inc., 1995.
2. H. Brittain, Polymorphism in Pharmaceutical Solids, Marcel Dekker, Inc., 1999.
3. S.R. Byrn, R.R. Pfeiffer and J.G. Stowell, Solid State Chemistry of Drugs, Second Edition, SSCI, Inc.,1999.
4. E.F. Fiese and T.A. Hagen, “Preformulation”, Chapter 8 in the Theory and Practice of Industrial Pharmacy, Lea & Febiger, Philadelphia, 1986.
5. M. Gibson, Pharmaceutical Preformulation and Formulation, HIS Health Group, Englewood, CO, 2001.
6. L.J. Ravin and G.W. Radebaugh, “Preformulation”, Chapter 75 in Remington’s Pharmaceutical Sciences, 18th edition, Mack Publishing Company, Easton, Pennsylvania, 1990.
7. W.Q. Tong, “Preformulation Aspects of Insoluble Compounds” in Water Insoluble Drug Formulation, Edited by R. Liu, Interpharm Press, 2000.
8. J. Wells, Pharmaceutical Preformulation, Ellis Horwood Limited, 1988.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






