

 About Authors:
About Authors:
Dr. HV Chavda1*, Ms. SK Patel1, Dr. CN Patel1,
1Shri Sarvajanik Pharmacy College,
Nr. Arvind Baug, Mehsana, Gujarat-384001, India.
*hvchavda@sspcmsn.org
ABSTRACT
Background: In present investigation an attempt has been made to formulate a fast dispersible formulation of aceclofenac using three different superdisintegrants. The role of superporous hydrogel particle as a superdisintegrant was checked for its further future applications. Materials and Methods: The selection of superdisintegrants viz. poly (Acrylamide-co-Acrylic acid) superporous hydrogel particles, cross carmellose sodium and starch 1500 were done using the simplex lattice design. Tablet formulations were prepared using direct compression technique and evaluated for hardness, weight variation, friability, drug content, dispersion time, wetting time, water absorption ratio and in vitro drug release studies. Results and Discussion: The dispersion time of all formulation showed less than 21 second. Superporous hydrogel particles did not showed significant improvement in dispersion time compared cross carmellose sodium and starch 1500. The in vitro drug release from batch F7, tablets containing equal proportions of superdisintegrants showed fast dispersion and fast release compared to other batches. Batch F7 was stable for the period of six months at 40 oC / 75 %RH. Conclusions: Combination of three superdisintegrants was more suitable in fast dispersible tablet formulation of aceclofenac. The superporous hydrogel particles showed equivalent effect as a superdisintegrant, however process complexity of its preparation suspects its role or an option as a superdisintegrant in fast dispersible or immediate release formulations.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-1628
INTRODUCTION
Aceclofenac is a derivative of the diclofenac group of non-steroidal anti inflammatory drug that exhibits analgesic and anti-inflammatory activities. The mode of action of Aceclofenac is based on the inhibition of prostaglandin synthesis directly. It has less gastrointestinal complications.[1,2] It is considered to be the first-line drug in the symptomatic treatment of rheumatoid arthritis, osteoarthritis and ankylosing spondylitis. [3] Aceclofenac can be administered twice daily as 100mg orally in the treatment of rheumatoid arthritis. Geriatric patients may have difficulty in swallowing and chewing the tablets resulting in patient non compliance and ineffective therapy. [4] Fast dispersible drug delivery system offers a solution for patients having difficulty in swallowing the formulations. For poorly soluble Aceclofenac, the rate of absorption is often controlled by the rate of dissolution. The dissolution of a drug can also be influenced by disintegration time of the tablets. Faster disintegration of tablets delivers a fine suspension of drug particles resulting in a higher surface area and faster dissolution.
In this investigation, attempts have been made to develop fast dispersible tablets of aceclofenac using various superdisintegrants.
MATERIALS AND METHOD
Materials
Aceclofenac and Starch 1500 were generous gift from Cadila Healthcare Ltd. and Claris Life-Sciences, Ahmedabad, respectively. Microcrystalline cellulose,mannitol, croscarmellose sodium, acrylic acid , N,N’-Methylene-bis-acrylamide, Span 80, ammonium persulphate, and N,N,N’,N’-Tetramethylethylenediamine were purchased from SD Fine Chem. Ltd, Mumbai, India. Acrylamide was obtained from Burgoyne Burbidges and Co. Pvt. Ltd., Mumbai, India. Double distilled water and 0.1N HCL were prepared in laboratory. All other chemicals used were of analytical grade and used as obtained.
Preparation of Fast dispersible Tablets
Fast dispersible tablets containing 100 mg of aceclofenac were prepared by direct compression method. The drug and additives were passed through 80# sieve, and mixed thoroughly by geometric mixing followed by addition of magnesium stearate and talc. After evaluation of powder blend the tablets were compressed using 7 mm flat faced round shaped punch. The composition for single tablet is shown in Table 1. Here three different superdisintegrants were taken and their proportion was set using a simplex lattice design. Superdisintegrants used were cross carmellose sodium, starch 1500 and Poly (Acrylamide-co-acrylic acid) superporous hydrogel particles. Superporous hydrogel particles were prepared as reported in our previous studies. [5-7]
Table 1: Composition of single fast dispersible tablet of aceclofenac
|
Ingredients (mg/tablet) |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
|
Aceclofenac |
100 |
100 |
100 |
100 |
100 |
100 |
100 |
|
Microcrystalline cellulose |
75 |
75 |
75 |
75 |
75 |
75 |
75 |
|
Mannitol |
37.5 |
37.5 |
37.5 |
37.5 |
37.5 |
37.5 |
37.5 |
|
Superporous Hydrogel Particle |
30 |
- |
- |
15 |
- |
15 |
10 |
|
Cross Carmellose Sodium |
- |
30 |
- |
15 |
15 |
- |
10 |
|
Starch 1500 |
- |
- |
30 |
- |
15 |
15 |
10 |
|
Talc |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
|
Magnesium Stearate |
2.5 |
2.5 |
2.5 |
25 |
2.5 |
2.5 |
2.5 |
|
Total Weight |
250 |
250 |
250 |
250 |
250 |
250 |
250 |
Evaluation of Tablets
The prepared tablets were evaluated for hardness, weight variation, thickness, friability, drug content, wetting time and dispersion time. For each formulation, the hardness (5 tablets) and friability (10 tablets) were determined using the Pfizer type hardness tester (Janki Impex, Ahmedabad) and the Roche friabilator (Electrolab, India), respectively. To study weight variation 20 tablets of each formulation were weighed and the test was performed according to Indian Pharmacopoeia 2007. For estimation of drug content, 20 tablets were crushed, and the aliquots of powder equivalent to 100 mg of drug were extracted in phosphate buffer pH 7.4. The solutions were passed through 0.45 µm membrane filter and after suitable dilutions the absorbance was measured at 274 nm using the UV-1800 UV/Vis Double Beam Spectrophotometer (Shimadzu, Japan).
Wetting Time and Water Absorption Ratio
For wetting time measurement a piece of tissue paper folded twice was placed in a Petri dish containing 6 ml of distilled water. A tablet was placed on the tissue paper and small amount of amaranth powder was placed on upper surface of tablet. The time required for development of a red color on the upper surface of the tablet was recorded as wetting time. [8] Three tablets from each batch were used to get an average value.Then wetted tablet was weighed and percentage of water absorption was determined using the equation 1:
R= (Wb – Wa) / Wa x 100 (1)
where, Wa is weight of tablet before water absorption, Wb is weight of tablet after water absorption, and R is water absorption ratio. [9]
In Vitro Dispersion Time
The in vitro dispersion time test was carried out by dropping a tablet in 10 ml phosphate buffer pH 7.4 at 37±0.5°C. The time taken for complete dispersion of the tablet was measured in seconds. [10]Three tablets from each batch were used to get an average value.
In Vitro Drug Release Studies
The release rate of aceclofenac from tablet(n = 3) was determined using USP XXIV Dissolution Testing Apparatus II (dissolution tester model TDT-08 L, Electrolab, India) at 100 rpm. The dissolution test was performed using 900ml of phosphate buffer pH 7.4 maintained at 37 ± 0.5 oC. At the regular time interval of 5 min aliquots (5 ml) were withdrawn from the dissolution apparatus for 1 hr; and the fluid removed was replaced with fresh dissolution medium, immediately. The aliquots were filtered and diluted to suitable concentrations with dissolution medium. The absorbance of these solutions was measured at 274 nm using UV-1800 UV/Vis Double Beam Spectrophotometer. The cumulative percentage of drug release was calculated using an equation obtained from a standard curve.
Stability Studies
The optimized batch was kept in airtight containers and stored in stability chamber (TH-90S, Thermolab, India) at 40 oC / 75 %RH for six months. [11] Results for in vitro dissolution studies obtained after six months were compared with the data obtained at the time of preparation. The similarity factor (f2) was applied to study the effect of storage on optimized batch.The f2 value is calculated from the equation 2:
n
f2 = 50 x log {[1 + (1/n) ∑ |Rj – Tj |2 ]-0.5 x 100} (2)
j = 1
Where, n is the number of dissolution time points and Rj and Tj are the percent dissolved of the reference product and test product at each time point j, respectively.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
RESULTS AND DISCUSSIONS
Evaluation of Tablets
The evaluation parameters for tablets are shown in Table 2. The hardness and percentage friability of the tablets of all batches ranged from 3.0 ± 0.07 to 3.4 ± 0.11 kg/cm2 and 0.84 ± 0.06 to 0.95 ± 0.05, respectively. The average percentage deviation in weight variation test of 20 tablets of each formulation was less than ± 5%. Drug content was found to be uniform among different batches of the tablets and ranged from 97.38 ± 0.19 to 99.15 ± 1.59.
Table 2: Evaluation parameters-I for aceclofenac tablets
|
Batch |
Hardnessa (kg/cm2) |
Friabilityb (%) |
Deviation in Weight Variationc (%) |
Drug Contentb (%) |
|
F1 |
3.0 ± 0.07 |
0.86 ± 0.05 |
3.45 ± 0.06 |
97.38 ± 1.39 |
|
F2 |
3.2± 0.14 |
0.84 ± 0.06 |
3.47 ± 0.05 |
98.62 ± 1.64 |
|
F3 |
3.1± 0.15 |
0.95 ± 0.05 |
3.38 ± 0.06 |
97.67 ± 1.92 |
|
F4 |
3.5± 0.08 |
0.93 ± 0.04 |
3.42 ± 0.07 |
98.99 ± 1.46 |
|
F5 |
3.0± 0.07 |
0.86 ± 0.06 |
3.48 ± 0.09 |
98.25 ± 1.71 |
|
F6 |
3.4± 0.11 |
0.89 ± 0.07 |
3.39 ± 0.06 |
99.15 ± 1.59 |
|
F7 |
3.2± 0.13 |
0.92 ± 0.07 |
3.43 ± 0.08 |
97.45 ± 1.84 |
a Mean ± SD, n = 5
b Mean ± SD, n = 10
c Mean ± SD, n = 20
Wetting Time and Water Absorption Ratio
Wetting time and water absorption ratio are shown in Table 3. Wetting time and percentage water absorption ratio were ranged from 56.33± 1.00 to 59.67 ± 1.15 second and 96.00 ± 1.00 to 98.17 ± 1.26 %.
Table 3: Evaluation parameters-II for aceclofenac tablets
|
Batch |
Dispersion time* (sec) |
Wetting time* (sec) |
Water Absorption Ratio* (%) |
Drug release in 5 min* (%) |
|
F1 |
16.67± 1.00 |
56.67± 3.06 |
98.10± 1.35 |
41.20± 3.04 |
|
F2 |
19.00± 1.53 |
59.67± 1.15 |
97.33± 1.53 |
46.93± 3.51 |
|
F3 |
16.00± 1.00 |
58.67± 2.08 |
96.00± 1.00 |
51.39± 3.02 |
|
F4 |
20.33± 1.15 |
57.33± 2.31 |
97.33± 1.53 |
52.38± 1.63 |
|
F5 |
18.33± 0.58 |
58.33± 1.53 |
98.17± 1.26 |
57.39± 2.21 |
|
F6 |
15.67± 0.58 |
56.33± 1.00 |
96.33± 1.53 |
58.22± 2.25 |
|
F7 |
15.33± 1.15 |
57.33± 0.58 |
97.00± 1.00 |
64.93± 2.51 |
In Vitro Dispersion Time
All batches were found to be fast dispersible as they all dispersed in just 21 second as shown in Table 3. Dispersion time was ranged from 15.33 ± 1.15 to 20.33 ± 1.15 second. Batch F7 was found to be fast dispersible formulation. Superdisintegrants used alone, Batch F1-F3, and in combination of two, Batch F4-F6, there was no significant difference in dispersion time. The dispersion time from tablets containing equal proportions of superdisintegrants showed fast dispersion, but no significant difference was there. Even though the dispersion time was very short, this is important in case of fast dispersible formulation, combination of superdisintegrants didn’t impart any significant change in dispersion time.
In Vitro Drug Release Studies
The drug release profiles from the prepared batches containing different proportions of selected superdisintegrants are shown in Figure 1. Batch F7 was able to provide the fast release 64.94 % of drug within 5 min. All batches showed also showed fast release and release more than 41 % of OMZ within 5 min. In case of superdisintegrants used alone, Batch F1-F3, there was slow drug release compared to in combination there of. Here superporous hydrogel particles showed less release compared to other two. When superdisintegrants were taken in combination of two, Batch F4-F6, the synergistic effect on drug release was observed, as drug release was faster compared superdisintegrants alone. The in vitro drug release from tablets containing equal proportions of superdisintegrants viz. superporous hydrogel particles, cross carmellose sodium and starch 1500, showed fast dispersion as well as fast release compared to all other batches. It could be possible that in combination of three superdisintegrants the synergistic effect might be there, which accelerate the drug release.
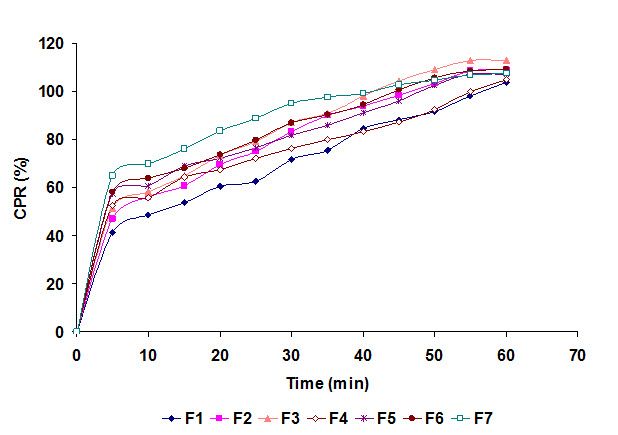
Figure 1: Drug release profiles from fast dispersible ACF tablets (n = 3)
Compared to routinely used superdisintegrants, superporous hydrogel particles, recently reported, played an important and equivalent role as a superdisintegrant. The superporous hydrogel particles have a high tendency of swelling and hence absorb the dissolution fluid and swells quickly; this imparts quick breakage of tablets and hence fast dispersion. The superporous hydrogel particles may be other option as a superdisintegrant or disintegrant for future formulations.
Stability Studies
Batch F7 was chosen for reference in order to calculate similarity factor (f2). After six months of applied stability conditions Batch F7 showed f2 value of 79.54. The stability studies revealed that there were no significant changes observed for in vitro dissolution studies after six months. Bath F7 was found to be stable for the period of six months at 40 oC / 75 %RH.
CONCLUSIONS
The simplex lattice design was used for the selection of superdisintegrants. The in vitro drug release from batch F7, tablets containing equal proportions of superdisintegrants showed fast dispersion and fast release compared to all other batches. Combination of three superdisintegrants was suitable for the fast dispersible tablet. The superporous hydrogel particles may be other option as a superdisintegrant in fast dispersible or immediate release formulations.
REFERENCES
1. Parfitt K. Analgesics Anti-inflammatory and antipyretics. In: Reynolds JEF, editor. Martindale: The complete drug reference. 32nd ed. Massachusetts: 1999. P. 2-12.
2. British Pharmacopoeia. The Stationary office, MHRA, British Pharmacopoeial Commission office. London: 2005. p. 1.
3. Theodore WR, Alan SN, Taylor P, Gilman AG, editors. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 8th ed. New York: McGraw-Hill; 1991: 1484.
4. Bhardwaj S, Jain V, Jat RC, Mangal A, Jain S. Formulation and evaluation of fast dissolving tablet of aceclofenac. Int J Drug Del. 2010; 2: 93-7.
5. Chavda HV, Patel CN, Prajapati ST, Patel CV. A Newer Formulation Approach Based on Superporous Hydrogel Particles for Gastroretentive Drug-Delivery System: Preparation and In Vitro Evaluation. Inventi Impact: NDDS. 2010; 1(1): Article ID: 2010nd70a (Published on Web 15/07/2010; inventi.in).
6. Chavda HV, Patel CN, Karen HD. Preparation and Characterization of Chitosan-Based Superporous Hydrogel Composite. J Young Pharm. 2009; 1: 199-204.
7. Chavda HV, Patel CN. Preparation and Characterization of Swellable Polymer-based Superporous Hydrogel Composite of Poly (Acrylamide-co-Acrylic Acid). Trends Biomat Artif Organs. 2010; 24: 83-9.
8. Yonnobu Y, Hisakazu S, Kazumi D, Akinobu O. Preparation and evaluation of a compressed tablet rapidly disintegration in the oral cavity. Chem Pharm Bull. 1996; 44: 2121-7.
9. Garala KC, Ekshinge VB, Jarag RJ, Shinde AJ. Fast-disintegrating aceclofenac tablets: formulation development using simplex lattice design. Thai J Pharm Sci. 2008; 32: 77-81.
10. Mohapatra A, Parikh RK, Gohel MC. Formulation, development and evaluation of patient friendly dosage forms of metformin, Part-I: Orally disintegrating tablets. Asian J Pharm. 2008; 2: 167-71.
11. Mathews BR. Regulatory aspects of stability testing in Europe. Drug Dev Ind Pharm. 1999; 25: 831-56.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






