

 About Authors: V.P. Patel*, M.C. Gohel, R.K. Parikh
About Authors: V.P. Patel*, M.C. Gohel, R.K. Parikh
*V. P. Patel
M.Pharm, Assistant Professor, R. K. College of Pharmacy, Rajkot, Gujarat, India.
Work was carried out at: Department of pharmaceutical technology,
L. M. College of pharmacy, Ahmedabad, Gujarat, India
M. C. Gohel
Professor and Principal, Department of pharmaceutical technology,
L. M. College of pharmacy, Ahmedabad, Gujarat, India
R. K. Parikh
Professor, Department of pharmaceutical technology,
L. M. College of pharmacy, Ahmedabad, Gujarat, India
Abstract:
With the introduction of combinatorial chemistry and high throughput screening, the properties of new chemical entities shifted towards higher molecular weight and increasing lipophilicity that results in decreasing aqueous solubility. It is not surprising that many drug candidates have poor water solubility since the initial selection of drug candidates are based on activity alone. Other physiochemical and biopharmaceutical properties such as permeability, biopharmaceutics and metabolism are rarely considered during the selection process. The aim of this study was to increase dissolution rate of felodipine poorly soluble drugs from BCS class II by complexation process. Complexation was prepared using β cyclodextrine. Different techniques were employed for preparation of complexation with β cyclodextrin like physical mixture, cogrinding, kneading technology, solvent coevaporation.The physical properties of the prepared solid mass of felodipine was characterized by in vitro dissolution studies, UV- spectroscopy, Fourior transform infrared spectroscopy, differential scanning calorimetry (DSC) and X-ray powder differaction spectroscopy. Additionaly, phase solubility studies were performed to support the in vitro dissolution study. The results of Fourior transform infrared spectroscopy shows the compatibility of drug with cyclodextrin, while differential scanning calorimetry (DSC) and X-ray powder differaction spectroscopy showed the confirmation of complexation of β cyclodextrin with felodipine.
[adsense:336x280:8701650588]
INTRODUCTION:
Felodipine is a potent dihydropyridine with almost exclusive action on vascular smooth muscle especially peripheral arterioles. In pharmaceutical science, it is widely accepted for its tolerant antihypertension and antianginal properties since it is a calcium antagonist compound (calcium channel blocker)[1]. Specifically, FEL is a compound with strong lipophilic character appearing as a crystalline powder practically insoluble in aqueous solutions[2-3].The terminal elimination half life of felodipine is reported to about 11 to 16 hours following oral administration of an immediate release preparation, but longer with a modified release formulation. [4]
Cyclodextrins (CDs) are a group of structurally related cyclic oligosaccharides that are formed by enzymatic cyclization of starch. The three most common naturally occurring CDs are a-cyclodextrin (aCD), b-cyclodextrin (bCD) and g-cyclodextrin (gCD) consisting of six, seven and eight (a-1,4)-linked a-D-glucopyranose units, respectively. The CD molecules are cone-shaped with a somewhat hydrophobic central cavity and hydrophilic outer surface. They are capable of forming inclusion complexes with many drugs by taking up a whole drug molecule, or more frequently, some hydrophobic part of it, into the cavity. [5]
MATERIALS AND METHODS:
Materials: Felodipine, Beta Cyclodextrin.
Methodology:
Phase solubility study:
Phase solubility study for felodipine was performed as described by Higuchi and Connors. [6] Excess amount of felodipine was added into aqueous solution containing carriers at various concentrations and shaken for 96 h at room temp on a shaker (time period of 96 hr was previously determined for achievement of felodipine equilibilium) and protected from light to equilibrate.After equilibilium, the suspensions were filtered through 0.45 μm Millipore membrane filters. The first 15% of the filtrate was discarded to avoid any potential loss of the drug, because of absorption by the filter until and the subsequent filtrate was collected. The filtrate was appropriately diluted by methanol and the concentration of the felodipine in the filtrate was determined by UV spectrophotometer (UV-1601PC, Shimadzu) at 362 nm. Earlier experiments showed that the presence of different carrier did not interfere with the assay at the concentration employed.Solubility measurements were performed in triplicate.
Methods of preparation: [7]
Preparation of physical mixture (PM): The physical mixture of FDP with β cyclodextrin was prepared in 1:8, 1:10 and 1.12 geomatrix ratiosby means of spatula for 20 minutes and passed through 80 meshscreens.
Preparation of cogrinding product (CG): For co-grinding product, FDP with β cyclodextrin were mixedin 1:8, 1:10 and 1.12 geomatrix ratioand triturated in glass mortar pestle for 20 minutes and passed through 80meshscreen.
Preparation of kneaded product (KN): The kneaded product was prepared by addition of drug into the paste of cyclodextrin, 1:8, 1:10 and 1.12 geomatrix ratio and triturating the resultant mixture till cracking sound appear.The resultant mass was dried at room temp. and pass through 80 meshscreen.
Preparation of co-evaporated product (CE): For the preparation of co-evaporated product, an aqueous solution of cyclodextrin was added to a methanolic solution of felodipine to get 1:8, 1:10 and 1.12 geomatrix ratiosof drug and cyclodextrin. The resulting mixture was stirred for 1 hour and evaporated at 45-50°c until nearly dry. The resulting mass was allowed to dry at room temp for two consecutive days. These dried mass pulverized and pass through 80 mesh screen.
Table 1: Formulations Batches of complexation of felodipine with β cyclodextrin
|
Methods of preparation |
Type of polymer |
Drug to polymer ratio |
Batch code |
|
Physical Mixture |
β cyclodextrin |
1:8 |
F1 |
|
1:10 |
F2 |
||
|
1:12 |
F3 |
||
|
Cogrinding product |
β cyclodextrin |
1:8 |
F4 |
|
1:10 |
F5 |
||
|
1:12 |
F6 |
||
|
Kneaded product |
β cyclodextrin |
1:1.5 |
F7 |
|
1:2 |
F8 |
||
|
1:2.5 |
F9 |
||
|
Co-evaporated product |
β cyclodextrin |
1:8 |
F10 |
|
1:10 |
F11 |
||
|
1:12 |
F12 |
Characterization:
UV Spectroscopy:UV spectra were obtained on a UV spectrophotometer, Shimadzu with a wavelength range of 200 nm- 400 nm.
Fourier transform infrared (FTIR): Fourier transform infrared (FTIR) spectra were obtained on a spectrophotometer with a resolution of 2 cm -1 from 3500 to 600 cm-1. Pellets were prepared by gently mixing the sample with potassium bromide (1:100 ratio).
Differential scanning calorimetry (DSC): DSC measurements were made on a Pyris- calorimeter (Perkin-Elmer). Approximately 3–5 mg samples were accurately weighed into standard aluminium pans. An empty pan was used as reference. The samples were heated from room temperature to 290 ?C with scan rates 5?C/min. All DSC curves were normalized to a sample mass of 1 g. The instrument was calibrated using indium (melting point, 156.61 °C; enthalpy of fusion, 28.71 J, g–1).
X Ray diffraction: The physical state of felodipine in pure drug powder and in optimized mass was evaluated by powder X-ray diffraction. Powder X-ray diffraction (XRD) patterns were determined with an X-ray diffractometer employing Ni-filtered CuKα radiation source operating at 30 kVoltageand a current of 40 mA.
In vitro dissolution characterization: In vitro dissolution studies were carried out in 500 ml of 0.1 N hydrochloric acid containning 1 % w/v SLS using USP dissolution apparatus with an aggitation of 100 RPM. SLS was used to maintain sink condition. Powder sample equivalent to 10 mg of drug was filled in empty capsule shell and immersed in the dissolution medium. The dissolution sample (5 ml) withdrawn at different time intervals was filtered through a membrane filter (0.45 µm). This filtered sample was diluted sufficiently by 0.1 N HCL and assayed spectrophotometrically at 362 nm. Fresh medium was added to maintain a constant volume after each sampling. The dissolution were conducted in triplicate and the means of the absorbance were calculated. The cumulative percentage of drug release after 15 min, 30 min, 45 min, 60 min, 120 min as well as time required for 90% drug release is measured.
RESULTS AND DISCUSSIONS:
Phase solubility study:
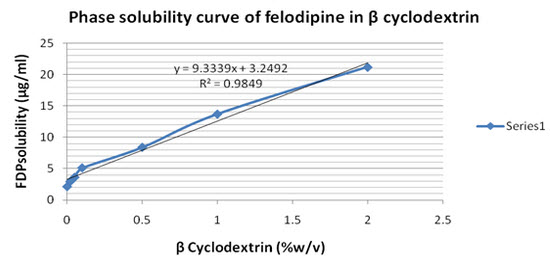
Figure 1 shows that solubility profiles of FDP in 0.1 N HCL influenced by carrier material concentration at room temp. The solubility curve with correlation coefficient squared values (r2) >0.95 (r2=0.95) was regarded as a straight line (AL type) (Higuchi and Connors, 1965).
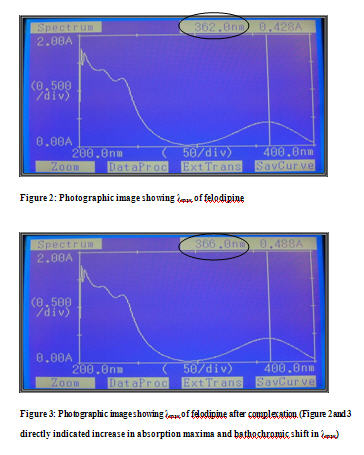
UV Spectroscopy: Formulation excipients selected on the basis of preliminary tests, which demonstrates no interference of these excipients with the λmax of FDP. [8] The complex formation with cyclodextrin alters the original UV absorption spectrum of the molecule usually bathochromic shift and or band broadening occurs. The shift of the UV absorption maxima upon complex formation may be explained by a particular shielding of the excitable electrons in the CD cavity. [9] [See Figure 2 and 3]
FTIR:
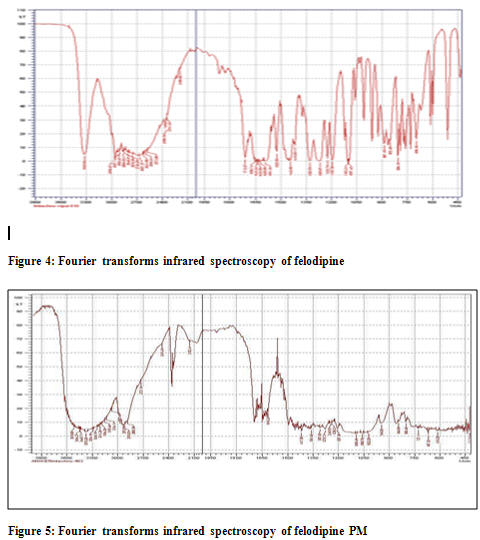
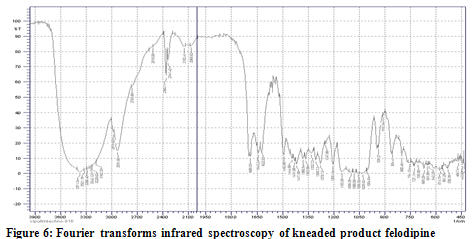
Complexation process of Felodipine with the beta cyclodextrin has been confirmed by the FT-IR spectroscopy. The FT-IR spectra of the inclusion complexes of the felodipine studied are significantly different from those of the corresponding physical mixtures and of felodipine alone. The PM of 1:1 ratio showed superimposed spectra of FDP and beta cyclodextrin which proves the compatibility of beta cyclodextrin with the FDP. In the FT-IR spectra of the inclusion compounds of Felodipine studied, the bands in the range 3050–3250 cm-1 with a maximum at 3070 cm-1, assigned to the stretching vibrations of the N–H bonds in the dihydropyridine ring are not clearly distinguished. The differences also appear in the band corresponding to the vibrations of carbonyl groups in the ester bonds. In the spectra of the felodipine the band occurs at 1690–1710 cm-1 while in the spectra of the corresponding inclusion complexes it is less broadened and % transmittance reduces. The differences also appear in the band corresponding to the amine group in the pyridine ring. In the spectra of the felodipine the band occurs at 3360-3380 cm-1 while in the spectra of the corresponding inclusion complexes it is much broadened. Some differences are also noted in the range 450-500 cm-1, band corresponding to the chlorine groups of phenyl which in case of inclusion complex shortened to a great extend. From these results, it can be speculated that drug-carrier hydrogen bonding existed in these solid binary system, causing reduced drug recrystallization.[10]
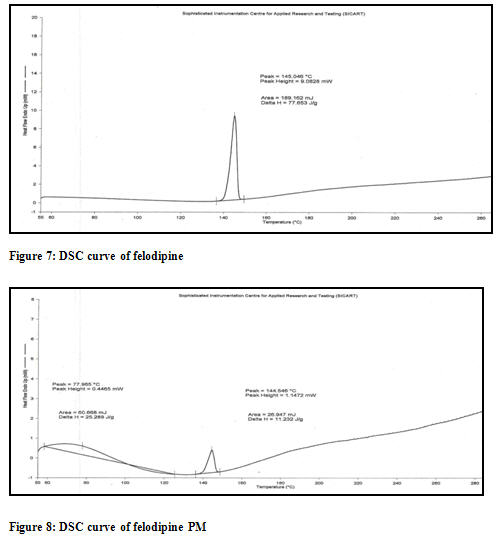
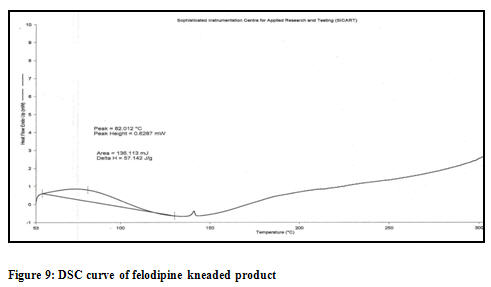
Differential scanning calorimetry (DSC): The DSC thermograms of felodipine, felodipine (PM), and the inclusion complexes of felodipine are illustrated into the Figure 7,8 and 9 respectively. Physical mixture showing almost similar identical melting endotherm and spectra are overlapable which proves the compatibility of used excipients [Figure 8]. The formation of inclusion complexes is indicated by the disappearance of peak or significant change in area of the peak or shift of the endothermic peaks corresponding to the drug melting process [Figure 9]
X Ray diffraction:
The powder X-ray diffraction (PXRD) patterns of pure FDP, and Kneaded complex are shown in Figure 10 and 11.
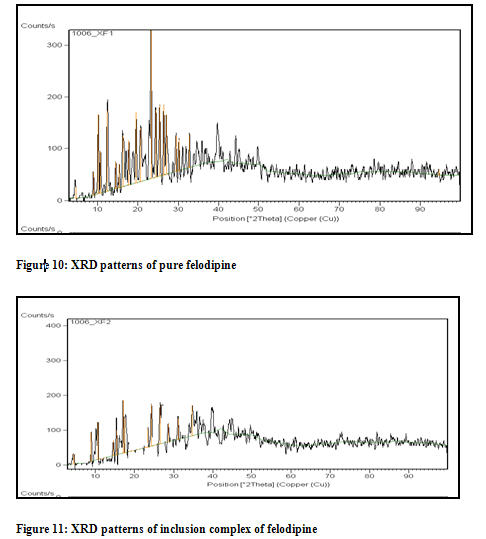
Felodipine presented a crystalline form as demonstrated by various intense diffraction peaks [Figure 10]. The crystalline peaks located at 4.6 º, 8.9 º, 10.1 º, 10.7 º, 12.4 º, 14.5 º, 15.3 º, 16.3 º, 17.7 º, 19.5 º, 20.6 º, 23.2 º, 24.4 º, 25.4 º, 26.4 º, 27 º, 29.4 º, 30.1 º, 32.7 º, and 94.5º (2 θ) corresponding to felodipine crystals were observed. Felodipine had a more intense peak at 10.1 º, 12.4 º, 16.3 º, 19.5 º, 20.6 º, 23.2 º, 24.4 º, 25.4 º, 26.4 º and 27 º in addition to relatively less intense peaks at 4.6 º, 8.9 º, 10.7 º, 14.5 º, 15.3 º, 17.7 º, 29.4 º, 30.1 º and 32.7 º. XRD patterns of kneaded binary systems showed reduced diffraction intensity as indicated in Figure 11. All these factors indicate some degree of amorphi-zation or inclusion complex formation FDP in cavity of cyclodextrin.
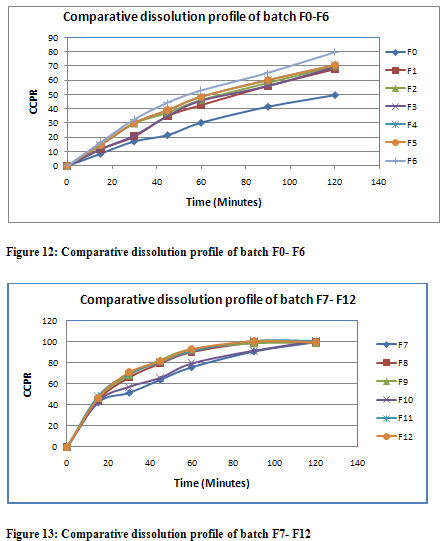
From the above figure 12 and 13, it was concluded that pure drug felodipine having very poor solubility and poor dissolution profile. The dissolution profile of F0 batch was suggested that only 49.5 % drug release occur after 2 hr in 0.1 N HCL and time required for 90 % drug dissolution is nearly about 4 hr, which suggests the poor dissolution characteristic of FDP. All the solid binary system prepared from physical mixture methods (F1 to F3) and cogrinding methods (F4 to F6) using β cyclodextrin not showing any significant improvement in dissolution characteristic of FDP. While in contrast other methods like kneading method (F7 to F9) and solvent co-evaporate methods (F10 to F12) produce significant enhancement in dissolution rate of FDP. However, the rate of dissolution enhancement by both kneading methods and solvent co-evaporated methods were nearly identical and showing almost similar dissolution profile. So, due to greater advantage of kneading method over solvent coevaporated method, kneading method was selected for further research work (see the table)
Based on phase solubility study and preliminary study, it was concluded that there was a linear enhancement in dissolution rate of FDP as the conc. of cyclodextrin increased. So, from the all the methods, kneading method was the optimized batch in 1:10 proportion of drug to β cyclodextrin which showing 90 % of drug release within 60 minutes.
CONCLUSION:
The aim of this work was to improve the dissolution rate of felodipine which is required for improving the dosage form characteristic and also to reduce fluctuation in dissolution profile as well as for better invivo characterization. Many techniques are known to improve dissolution rate of poorly soluble drugs amongst these preparation of physical mixture, co-grinding techniques, kneading techniques and solvent co-evaporated techniques are selected in this research work because ofits ease of preparation, ease of optimization, and reproducibility. Amongst these techniques kneading method and solvent co-evaporated methods found to be more effective than previous two techniques. Now, in industry due to the concept of green formulation, always the formulation without solvent is encourage by industry person because the sometimes presence of solvents creates volatile impurities. With consideration of concentartion of cyclodextrin and the concept of green formulation, Batch F8 was considered to be the optimised batch showing significant improvement in dissolution profiles that utilized 1:10 ratio of drug and beta cyclodextrin using kneading techniques.
REFERENCES:
1. Edgar B., Collste P., Haglund K., & Regardh CG. Pharmacokinetics and haemodynamic effects of felodipine as monotherapy in hypertensive patients. Clinical and Investigative Medicine, 1987; 10:388-394.
2. Abrahamsson B., Johansson D., Torstensson A., & Wingstrand K. Evaluation of solubilizers in the drug release of hydrophilic matrix extended-release tablets of felodipine. Pharmaceutical Research, 1994; 11 Suppl 8:1093 – 1097.
3. Evangelos Karavas, Georgios Ktistis, Aristotelis Xenakis, Emmanouel Georgarakis,. Miscibility Behavior and Formation Mechanism of Stabilized Felodipine-Polyvinylpyrrolidone Amorphous Solid Dispersions. Drug Development and Industrial Pharmacy 2005; 31 Suppl 6:473–489.
4. Alfred G.G. The Pharmacological basis of Therapeutics, 10th edition, Mc Graw Hill: 2001; 858-860.
5. Thorsteinn Loftsson, Hafrun Frioriksdottir. The effect of water-soluble polymers on the aqueous solubility and complexing abilities of b-cyclodextrin. International Journal of Pharmaceutics 1998; 163:115–121.
6. Higuchi T. and Connors KA. Phase solubility techniques. Advances in analytical chemistry and Instrumentation 1965; 4:117-212.
7. Mishra PR., Mishra M., Namdeo A., Jain NK. Review article: Pharmaceutical potential of cyclodextrin. International Journal of pharmaceutical science 1999; 61:193-198.
8. Rakesh P. Patel, Dhaval J. Patel, Dipen R. Bhimani and Jayvadan K. Patel. Physicochemical characterization and dissolution study of solid dispersions of furosemide with polyethylene glycol 6000 and polyvinylpyrolidone K 30. Dissolution technologies 2008; 2:17-23.
9. UV Szejtli J., Gerlozya, Fonagy A, Akademiai Kiado. Cyclodextrin and their inclusion complexation. Arzenimittelforsch 1980; 30:808.
10. Silverstein RM. Spectrometric Identification of organic compounds, 4 th Edition, John Wiley and Sons, New york, 1981, 112.
Reference ID: PHARMATUTOR-ART-1012
FIND OUT MORE ARTICLES AT OUR DATABASE






