

About Authors:
Garg Pushpendra, Nanda Arun
Department of Pharmaceutical Sciences
Maharshi Dayanand University, Rohtak-124001
Haryana, India.
ABSTRACT
The objective of the present study was to improve the dissolution rate of gliclazide, a poorly water soluble (BCS class-II) drug by solid dispersion technique using a water soluble carrier, poloxamer 407. The solid dispersions were prepared by fusion method using different concentrations of carrier and the prepared systems showed an enhancement in dissolution. Solid dispersions were characterized with Differential scanning calorimetry, X-ray diffraction, Fourier transform infrared spectroscopy, scanning electron microscopy and dissolution analysis. The study revealed that enhanced dissolution of gliclazide from solid dispersion was due to a decrease in crystallinity of drug and also due to dissolution of gliclazide in molten form of solid dispersion. In conclusion preparation of gliclazide dispersion with meltable hydrophilic polymer could be a promising approach to improve the dissolution rate.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1124
INTRODUCTION
The therapeutic efficacy of a drug product intended to be administered by the oral route depends primarily on its absorption by the gastrointestinal route. It is well established that the active ingredient in a solid dosage form must undergo dissolution before it is available for absorption from the gastrointestinal tract. Many potential drug candidates are characterized by a low oral bioavailability. Often, poor drug dissolution and solubility rather than limited permeation through the epithelia of the gastrointestinal tract are responsible for low bioavailability of orally taken drugs. Therefore, the solubility of a drug is an important factor in determining the rate and extent of its absorption (1) and an enhancement in dissolution rate is important to attain suitable blood-levels of BCS class II drugs (low solubility and high permeability). Gliclazide is an example of class II drug which is second generation hypoglycemic sulfonylurea useful in the treatment of type 2 diabetes mellitus (2). Current literature shows that the drug has good tolerability, low incidence of hypoglycemia, low rate of secondary failure, inhibits platelet aggregation and increases fibrinolysis (3,4). For these reasons, gliclazide appears to be a drug of choice in long term sulfonylurea therapy for the control of type 2 diabetes mellitus (5). Gliclazide exhibits poor aqueous solubility and unsatisfactory dissolution rate. Improvement in its solubility and dissolution rate is the primary reason for this study, as this improvement could be achieved by the use of water soluble polymers based on solid dispersion (SD) technology (6). Many water-soluble carriers have been employed for preparation of SD of poorly water soluble drugs. The most common are polyethylene glycols (7), polyvinyl pyrrolidone (8), lactose (9), β-cyclodextrin (10), hydroxy propyl methyl cellulose (11). Recently poloxamer, a group of block co-polymer non- ionic surfactants, have attracted considerable attention (12). Poloxamer block copolymers have been introduced in the late 1950s and since then they have been proposed for diverse pharmaceutical applications (13-15). They are listed in the US and European Pharmacopoeia (16). This group of copolymers consists of ethylene oxide (EO) and propylene oxide (PO) blocks arranged in a triblock structure EOx-POy-EOx. Their chemical formula is HO[CH2-CH2O]x [CH(CH3)-CH2O]y [CH2- CH2O]xOH, y is higher than 14. Registered trademarks of these copolymers (e.g., Pluronic®, Synperonic® or Tetronic®) cover a large range of liquids, pastes and solids. They are synthesised by sequential polymerisation of PO and EO monomers in the presence of sodium hydroxide or potassium hydroxide (17). These copolymers have amphiphilic properties characterised by their HLB values (hydrophilic-lipophilic balance), which highly depend on x and y values. By varying the values of these parameters, size, lipophilicity and hydrophilicity can be easily modified. Poloxamer 407 (European Pharmacopoeia 5th Edition), principally available in the registered trademark of Pluronic F127® (BASF laboratories, Wyandoote, USA) and Synperonic F127® (ICI laboratories, Wilton, UK), has a molecular weight of about 12,600 (9,840-14,600) (18), x and y are equal to 95-105 and 54-60, respectively. Its HLB is 22 at 22ºC (17,19). FDA guide has presented poloxamer 407 as an inactive ingredient for different types of preparations (e.g., I.V., inhalation, oral solution, suspension, ophthalmic or topical formulations) (16). The rationale of the present study was to investigate the use of poloxamer 407 for the preparation of solid dispersions with the objectives of improving dissolution rate of gliclazide. The possible interactions between gliclazide and poloxamer 407 in both solid and liquid states were investigated. Interaction in solid state was investigated by Fourier-transform infrared (FT-IR) spectroscopy, X-ray diffraction (XRD) analysis, Differential scanning calorimetry (DSC) and Scanning electron microscopy (SEM). Interaction in solution was studied by dissolution experiments.
[adsense:468x15:2204050025]
MATERIALS AND METHODS
Materials A gift sample of gliclazide was received from Synmedic laboratories (Faridabad, India). Poloxamer 407 was received as a gift sample from BASF chemicals (Mumbai, India). Double-distilled water was used throughout the study and all the other chemicals used were of analytical grade.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Method
Preparation of the solid dispersions by Fusion method Solid dispersions of gliclazide at five mass ratios (1:0.5, 1:1, 1:2, 1:4 and 1:8) were prepared with poloxamer by the melt or fusion method (20, 21). Poloxamer 407 was placed in a porcelain dish and allowed to melt by heating up to 60 °C. To the molten mass, an appropriate amount of gliclazide was added and stirred constantly untill homogenous dispersion was obtained. For rapid solidification, the resultant solution was cooled in an ice bath and stored in desiccator for 24 h. It was then scrapped, pulverized and passed through a sieve. The prepared solid dispersions were then filled in glass bottles, sealed and stored in a desiccator till further use.
Preparation of Physical mixture
Physical mixture of gliclazide with poloxamer 407 in mass ratio (1:8) was prepared by light trituration for 2 minutes. The mixture was passed through a sieve. The prepared mixture was then filled in glass bottles, sealed and stored in a desiccator till further use.
Drug Content analysis
The drug content in each solid dispersion and physical mixture was determined by the high performance liquid chromatography (HPLC) (Agilent technology, 1200 series). Various steps followed are given below:-
Preparation of solvent mixture: Solvent mixture was prepared by mixing 45 volume of acetonitrile and 55 volume of water.
Preparation of stock solution: Dissolved 50 mg of gliclazide in 23 ml of acetonitrile in 50 ml volumetric flask, volume was made with water upto 50 ml. Various dilutions were made using solvent mixture.
Method followed:
Chromatographic system
Column: A stainless steel column 25 cm × 4.6 mm packed with octadecylsilane bonded to porous silica (5 µm).
Mobile phase: A mixture of 0.1 volume of triethylamine, 45 volume of acetonitrile and 55 volume of water.
Flow rate:1.0 ml per minute.
Wavelength:Spectrophotometer set at 235 nm.
Injection volume:A 20 µl loop injector.
In vitro Dissolution studies
The goal of dissolution testing is to ensure the pharmaceutical quality of the product, which means ability of manufacturing reproducibility, release properties and also the biopharmaceutical characteristics, such as rate of release and extent of absorption.The in-vitro dissolution tests were performed for the pure gliclazide, physical mixture, and solid dispersions using USP dissolution test apparatus type II (paddle type) (Lab India DF 8000). The dissolution medium (900ml) used was 0.1 N HCl maintained at 37± 0.5 0C. Aliquots of sample were withdrawn at a specified time intervals to assay the released drug by HPLC at 235 nm. Aliquots of samples were replenished with equal volume of dissolution medium.
Differential Scanning Calorimetry (DSC)
DSC studies were performed using Differential Scanning Calorimetry DSC Q10 9.0 build 275 model Water Ltd. Samples were scanned in a hermetically sealed aluminium pan with ramp 10 ºC/min from 25 ºC to 200 ºC. An atmosphere of nitrogen (flow rate 60 ml/min) was maintained during the experiment. An empty aluminum pan was used as a reference.
Fourier Transform–Infra Red Spectroscopic (FT-IR) Studies
FT-IR analysis provides a complete vibrational motion analysis of the molecule. Thus, the infrared spectrum can be used for molecules much as a fingerprint can be used for humans. FT-IR was done to check the compatibility of excepient amongst each other. The spectrums wererecorded in the wavelength region of 4000cm-1 to 500 cm-1. The procedure consists ofdispersing a sample (drug alone or mixtureof drug and excepient) in KBr andcompressing into discs by applyingpressure. The pellet of the drug and carriers was placed in sample holder and purged with nitrogen gas for 5 min. and the spectrums were obtained.
X-Ray Diffraction (X-RD) Studies
The crystalline state of different samples was evaluated with X-ray powder diffraction. The patterns were obtained at room temperature using a PW1710 X-ray Diffractometer with Cu as anode material and graphite monochromator, operated at a voltage of 35 kV, current 40 mA. The samples were analyzed in the 2θ angle range of 5–50°, and the process parameters were set as: scan step size of 0.02° (2θ) and scan step time of 0.5 s.
Shape and Surface Morphology
The morphology and appearance of selected solid dispersion SD-E and gliclazide, were examined by Scanning Electron Microscope (SEM)operated at an accelerating voltage of 15 kV. at various magnification. Samples were prepared by mounting powder on to a brass stub using graphite glue and coated with gold under vacuum before use.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
RESULTS
Drug content analysis Results depicted in table 1 shows that the drug concentration in physical mixtures and solid dispersions ranged between 98.4, 98.2, 98.6, 97.9 and 99.1, 98.7%, respectively.
Table 1. Drug content in physical mixtures and solid dispersions
|
Physical mixture & Solid dispersions (Drug to Poloxamer 407 mass ratio) |
Drug content (%)a |
|
Physical mixture (PM) ( 1:8) |
98.4±1.6 |
|
Solid dispersion 1 (SD-A) (1:0.5) |
98.2 ±1.2 |
|
Solid dispersion 2 (SD-B) (1:1) |
98.6±1.5 |
|
Solid dispersion 3 (SD-C) (1:2) |
97.9 ±1.8 |
|
Solid dispersion 4 (SD-D) (1:4) |
99.1±0.5 |
|
Solid dispersion 5 (SD-E) (1:8) |
98.7±1.8 |
aMean ± SD, n = 3
Dissolution studies Dissolution of the pure drug, physical mixture as well as solid dispersions were tested in 0.1 mol L-1 HCl (pH 1.2) for a period of 120 minutes. Dissolution of the pure drug was found to be 23.05 % in 60 minutes. In the first 60 minutes, cumulative drug release from physical mixture (1:8) was 45.93 %, while solid dispersions showed 39.28, 71.33, 77.78, 80.80, and 99.3 %, release. Results are presented in fig. 2. and table 2.
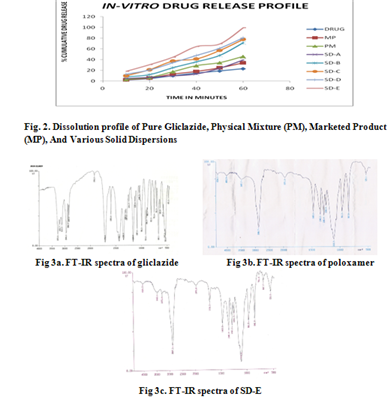
Table 2. In-Vitro dissolution data for Pure Gliclazide, Physical Mixture, Marketed Product, And Various Solid Dispersions
|
Time (min.) |
% CUMULATIVE DRUG RELEASE |
|||||||
|
Pure Drug |
MP |
PM |
SD-A |
SD-B |
SD-C |
SD-D |
SD-E |
|
|
10 |
1.84± 4.5 |
2.40±3.5 |
3.02±2.4 |
4.66±5.3 |
7.58±4.6 |
9.67±3.8 |
12.73±3.5 |
18.37±5.1 |
|
20 |
4.58±3.5 |
5.70±2.8 |
6.67±5.3 |
6.76±2.8 |
11.84±1.8 |
21.12±3.4 |
20.60±3.9 |
30.67±4.1 |
|
30 |
9.85±3.0 |
13.01±0.8 |
17.40±2.8 |
9.47±3.0 |
24.75±1.9 |
37.15±2.5 |
34.26±2.6 |
44.66±3.7 |
|
40 |
14.85±3.8 |
17.71±4.1 |
28.63±4.8 |
13.07±3.8 |
35.74±2.0 |
41.20±4.2 |
47.71±3.1 |
64.34±3.7 |
|
50 |
18.90±3.8 |
24.74±3.1 |
34.28±2.8 |
23.87±4.2 |
47.81±4.8 |
57.04±3.1 |
61.33±2.9 |
69.20±2.6 |
|
60 |
23.05±1.2 |
33.91±3.0 |
45.93±2.2 |
39.28±3.9 |
71.33±4.2 |
77.78±2.2 |
80.80±3.7 |
99.28±1.8 |
Possible mechanisms of increased dissolution rates of solid dispersions include a reduction of crystallite size, solubilization effect of the carrier, absence of aggregation of drug crystallites, improved wettability and dispersibility of the drug from the dispersion, dissolution of the drug in the hydrophilic carrier, drug conversion to amorphous state and finally, a combination of the mentioned mechanisms (22). The increased dissolution rate in these cases can thus be attributed to several factors, such as the solubilization effect of the carrier, conversion to amorphous state, and improved wettability of gliclazide.
Fourier transform infrared (FT-IR) spectroscopy The FT-IR spectrums of gliclazide, poloxamer 407 and solid dispersion (SD-E) are presented in figs. 3a-c. The spectrum of gliclazide showed a sharp concave curve at 1708.8 cm–1 for the carbonyl group. A symmetric stretching peak at 1164 cm–1 and an asymmetric stretching peak at 1350 cm–1 were detected for the sulphonyl group. A peak at 3272 cm–1 evidenced the amino group. The poloxamer 407 spectrum showed important peaks at 1109 cm–1 (C-O-C stretch) and at 2889 cm–1 (CH stretch). In this case, any sign of interaction would be reflected by a change in C=O, S=O and NH vibrations in the spectrum of solid dispersions, depending upon the extent of interaction. The spectra of solid dispersions did not indicate any well-defined interaction between gliclazide and poloxamer 407.
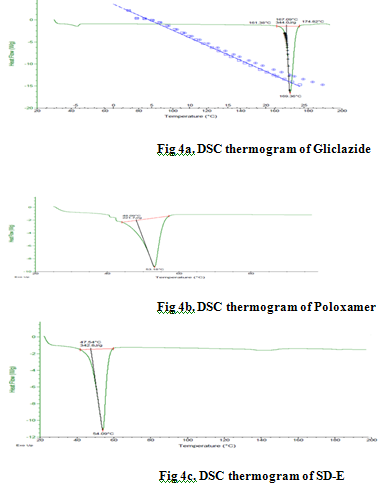
Differential Scanning Calorimetry (DSC) studies The DSC thermograms of gliclazide, poloxamer 407 and SD-E are presented in Figs. 4a-c. Gliclazide showed an endothermic reaction and its melting peak was at 167.39°C. The thermal behavior of poloxamer 407 exhibited a sharp but slightly broad endothermic peak at 49.09°C. and solid dispersion (SD-E) showed a single sharp melting peak 49.3°C, respectively. Complete disappearance of the gliclazide melting peak observed in SD-E was attributable to complete miscibility of the drug in the melted carrier.
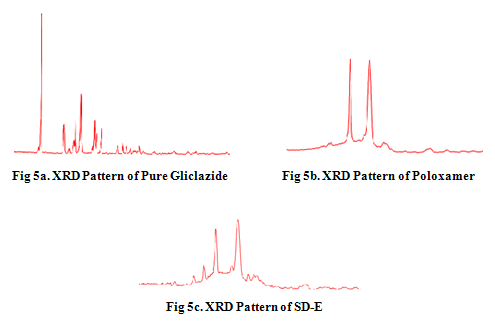
X-ray diffaraction studies The diffraction patterns of pure gliclazide, poloxamer 407 and solid dispersion (SD-E) (1:8) are shown in Figs. 5a-c. The diffraction pattern of pure gliclazide showed that the drug was of crystalline nature, as demonstrated by numerous distinct peaks. Gliclazide’s numerous diffraction peaks were observed at 2θ of 10.5, 15.0, 16.7, 17.0, 17.8, 18.1, 18.4, 20.8, 21.1, 22.0, and 26.2, etc. (fingerprint region), indicating the crystalline nature of gliclazide. Pure poloxamer 407 showed two peaks with highest intensity at 2θ of 19.07 and 23.15. Solid dispersion showed peaks of the drug; however, the intensity of the peaks was reduced when compared to that of the drug and hence absent. The results indicate that the drug in solid dispersion was amorphous as compared to the pure drug; hence the dissolution of the drug was improved. Poloxamer 407 peaks in solid dispersion were the same and just superimposed, which ruled out the possibility of chemical interaction between gliclazide and poloxamer 407.
Shape and Surface Morphology
The surface morphology studies revealed that the solid dispersion was closely compacted into small spherical form. Gliclazide existed in large crystalline particles of rather irregular size. On the contrary, the solid dispersions appeared in the form of spherical particles and the original morphology of components disappeared, which supported DSC and XRD data. These results demonstrated that gliclazide in solid dispersion was homogeneously dispersed into poloxamer at the molecular level.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
DISCUSSION
The increase in solubility of gliclazide was observed using hydrophilic polymer (poloxamer 407). Between the five ratios used, solid dispersion of drug:carrier (1:8) showed better effect on solubility of gliclazide compared to other ratios. The increase in solubility of gliclazide by poloxamer probably may be due to the formation of soluble complexes between water-soluble polymeric carrier and poorly soluble drug.
The solid state characteristics indicated decreased crystallinity of drug in the form of solid dispersion. The diffractograms of pure gliclazide exhibited a series of intense peaks, which were indicative of its crystallinity. Intensity of peak sharpness was reduced in solid dispersion compared to pure drug. An increase in the dissolution rate of gliclazide has been attributed to the hydrophilic nature of the polymer, which increased the wettability of the drug and also due to decrease in its crystallinity when prepared as a solid dispersion, which was confirmed by DSC and XRD results. In presence of poloxamer, drug had better wettability, hence the dissolution of drug was greater in the form of solid dispersion.
CONCLUSIONS
The solubility and dissolution rate of gliclazide can be enhanced in SDs with poloxamer 407. The solubilization effect of poloxamer 407, reduction of particle aggregation of the drug, absence of crystallinity, increased wettability and dispersibility, and alteration of surface properties of the drug particles may be responsible for the enhanced solubility and dissolution rate of gliclazide from its SDs and PM. DSC of SD-E does not indicate the presence of crystalline gliclazide, because gliclazide dissolved completely below its melting point. The absence of an endothermic peak of gliclazide in the DSC thermograms of SD-E with poloxamer 407 shows the conversion of gliclazide from the crystalline to the microcrystalline state. The FT-IR spectroscopic studies show the absence of any specific chemical interaction between gliclazide and poloxamer 407 in the solid state. It can be concluded that gliclazide SDs with poloxamer 407 provide a promising way to enhance its solubility and dissolution rate.
REFERENCES
1. Orienti I, Bigucci F, Luppi B, Cerchiara T, Zuccari G, Giunchedi P (2002) Polyvinylalcohol substituted with triethyleneglycolmonoethylether as a new material for preparation of solid dispersion of hydrophobic drugs. Eur J Pharm Biopharm 54: 229–233.
2. Reynolds JEF. (Ed.) Martindale1993. The Extra Pharmacopoiea XXX, 30th ed. The Pharmaceutical Press, London, p. 279- 280.
3. Dollery SC (1991) Therapeutic Drugs. Churchill Livingstone, London,.
4. Harrower AD (1994) Comparison of efficacy, secondary failure rate and complications of sulfonylurea. J Diabetes Complications 8: 201–203.
5. Palmer KJ, Brogden RN (1993) Gliclazide, an update of its pharmacological properties and therapeutic efficacy in NIDDM. Drugs 46: 92-125.
6. Ford JL (1986) The current status of solid dispersions. Pharm Acta Helv 61: 69-88.
7. Liu C, Liu C, Desai KGH (2005) Enhancement of dissolution rate of valdecoxib using solid dispersions with polyethylene glycol 4000. Drug Dev Ind Pharm 31: 4-10.
8. Hirasawa N, Ishise S, Miyata H (2003) An attempt to stabilize nivaldipine solid dispersion by the use of ternary systems. Drug Dev Ind Pharm 29: 997-1004.
9. Hirasawa N, Danij K, Haruna M, Otuska A (1998) Physicochemical characterisation and drug release studies of naproxen solid dispersions using lactose as a carrier. Chem Pharm Bull (Tokyo) 46: 1027-1030.
10. Rawat S, Jain SK (2003) Rafecoxib-β-cyclodextrin inclusion complex for solubility enhancement. Pharmazie 58: 639-641.
11. Okimoto K, Miyake M, Ibuki R, Yasumura M, Ohnishi N, Nakai T (1997) Dissolution mechanism and rate of solid dispersion particles of nivaldipine with hydroxypropylmethyl cellulose. Int J Pharm 159: 85-93.
12. Chen Y, Zhang G, Neilly J, Marsh K, Mawhinney D, Sanzgiri Y (2004) Enhancing the bioavailability of ABJ-963 using solid dispersion containing pluronic F-68. Int J Pharm 286: 69-80.
13. Koffi AA, Agnely F, Ponchel G, and Grossiord JL (2006) Modulation of the rheological and mucoadhesive properties of thermosensitive poloxamer-based hydrogels intended for the rectal administration of quinine. Eur J Pharm Sci 27(4): 328-335.
14. Dumortier G, Kateb N, Sahli M, Kedjar S, Boulliat A, Chaumeil JC (2006) Development of a thermogelling ophthalmic formulation of cysteine. Drug Dev Ind Pharm 32(1): 63-72.
15. Dumortier G, Zuber M, Barges N, Chast F, Dutertre H, Chaumeil JC (1994) Lacrimal and plasmatic kinetics of morphine after an ophthalmic delivery of three different formulations. Drug Dev Ind Pharm 20(7): 1147-1158.
16. Rowe R, Sheskey P, Owen S (2005) Pharmaceutical Handbook of Pharmaceutical Excipients, 5th ed., Pharmaceutical, London UK and American Pharmaceutical Association, Washington, USA.
17. Kabanov AV, Batrakova EV, Alakhov VY (2002) Pluronic block copolymers as novel polymer therapeutics for drug and gene delivery. J Control Release 82(2-3): 189-212.
18. Takats Z, Vekey K (2001) Qualitative and quantitative determination of poloxamer surfactants by mass spectrometry. Rapid Commun Mass Spectrum 15(10): 805-810.
19. Moghimi SM, Hunter AC (2000) Poloxamers and poloxamines in nanoparticle engineering and experimental medicine. Trends Biotechnol 18(10):412-420.
20. Bhanubhai NS, Haresh M, Shailesh A, Shah R, Vijay K (2006) Preparation and characterization of etoricoxib-polyethylene glycol 4000 plus polyvinylpyrrolidone K30 solid dispersions. Acta Pharm 56: 285–298.
21. Leuner C, Dressman J (2000) Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm 50: 47-60.
22. Aulton ME (2002) Pharmaceutics – The Science of Dosage Form Design, 2nd ed., Churchill Livingstone, London, p. 8.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Job Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






