

 About Authors:
About Authors:
Arshad Hala*, Prof. Rajesh Dholpuria, Nilesh sovasia
Seth G. L. Bihani S. D. College Of Technical Education,
Institute Of Pharmaceutical Sciences & Drug Research,
Gaganpath, Sri Ganganagar, Rajasthan 335001
*Arshad_hala@yahoo.com
ABSTRACT:
Over the past fifty years nuclear magnetic resonance spectroscopy, commonly referred to as NMR, has become the preeminent technique for determining the structure of organic compounds. Nuclear Magnetic Resonance (NMR) spectroscopy is an analytical chemistry technique used in quality control and research for determining the content and purity of a sample as well as its molecular structure. For example, NMR can quantitatively analyze mixtures containing known compounds. For unknown compounds, NMR can either be used to match against spectral libraries or to infer the basic structure directly. Once the basic structure is known, NMR can be used to determine molecular conformation in solution as well as studying physical properties at the molecular level such as conformational exchange, phase changes, solubility, and diffusion.Of all the spectroscopic methods, it is the only one for which a complete analysis and interpretation of the entire spectrum is normally expected. Although larger amounts of sample are needed than for mass spectroscopy, NMR is non-destructive, and with modern instruments good data may be obtained from samples weighing less than a milligram. To be successful in using NMR as an analytical tool, it is necessary to understand the physical principles on which the methods are based.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1542
* BASIC NMR PHENOMENON:
We are performing experiments on the nuclei of atoms, not the electrons.
Subatomic particles (electrons, protons and neutrons) can be imagined as spinning on their axes. The rules for determining the net spin of a nucleus are as follows;
- If the number of neutrons and the number of protons are both even, then the nucleus has NO spin.
- If the number of neutrons plus the number of protons is odd, then the nucleus has a half-integer spin (i.e. 1/2, 3/2, 5/2)
- If the number of neutrons and the number of protons are both odd, then the nucleus has an integer spin (i.e. 1, 2, 3)
The overall spin, I, is important. Quantum mechanics tells us that a nucleus of spin I will have 2I + 1 possible orientations. A nucleus with spin 1/2 will have 2 possible orientations. In the absence of an external magnetic field, these orientations are of equal energy. If a magnetic field is applied, then the energy levels split. Each level is given amagnetic quantum number, m.
[adsense:468x15:2204050025]
Nuclei with odd mass numbers have half-integer spin quantum numbers.
i.e. 13C, 1H, 31P are spin I = 1/2
17O is spin I = 5/2
Nuclei with an even mass number and an even charge number have spin quantum numbers of zero.
i.e. 12C
Nuclei with an even mass number and an odd charge number have integer spin quantum numbers.
i.e. 2H is spin I = 1
Electrons also have a spin quantum number of 1/2
* 1H-NMR SPECTROSCOPY: THEORY:
A spinning charge generates a magnetic field, as shown by the animation figure.1.
The resulting spin-magnet has a magnetic moment (μ) proportional to the spin.

Figure: 1
In the presence of an external magnetic field (B0), two spin states exist, +1/2 and -1/2. The magnetic moment of the lower energy +1/2 state is aligned with the external field, but that of the higher energy -1/2 spin state is opposed to the external field. Note that the arrow representing the external field north point.
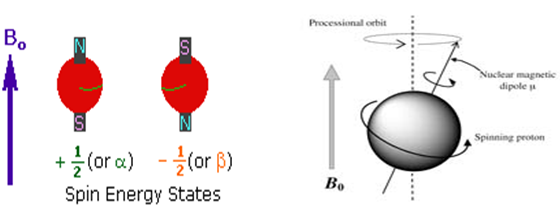
Figure: 2
This is a precessional motion and the top is said to be precessing around the vertical axis of the earths gravitational field.
* Precessional Frequency:
The spinning frequency of nucleus does not change but speed of precession changes. Precessional frequency (V1) isdirectly proportional to Strength of the External field (Bo) In an applied magnetic field like the proton precess at a frequency V1 which is proportional to the strength of applied field
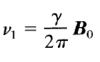
Bo = Strength of external magnetic field experienced by proton
γ = Magnetogyric ratio (Ratio between nuclear magnetic moment μ and nuclear angular momentum I)
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
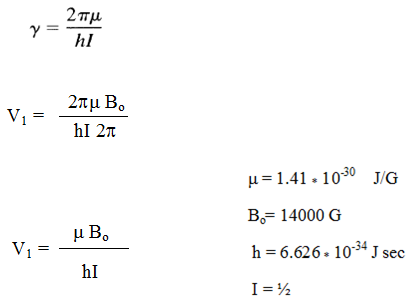
Protons will precess 60 million times per sec in 14000 G mag. field.
So proton will absorb at frequency of 60 MHz when 14000 Gauss external field is applied.
Similarly NMR frequency for various other nuclei can be calculated taking their respective μvalues.
E.g. 13C absorb at 15 MHz.
19F Absorb at 56 MHz when 14000 gauss external magnetic field is applied.
* Energy Transition:
The difference in energy between the two spin states is dependent on the external magnetic field strength, and is always very small. The following diagram illustrates that the two spin states have the same energy when the external field is zero, but diverge as the field increases. At a field equal to Bo a formula for the energy difference is given (remember I = 1/2 and μ is the magnetic moment of the nucleus in the field).
A proton in an external magnetic field of 1.4 tesla will precess at a frequency of 60 MHz and is capable of taking one of two orientation with respect to the axis of external field aligned or opposed i.e. parallel or anti parallel.
If proton is precessing in aligned orientation it can absorb energy and pass into the opposed orientation. It can also lose this extra energy and relax back into aligned position. The energy of a nucleus in the presence of a externally applied magnetic field is given by:
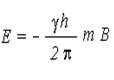
γ = Magnetogyric ratio (Ratio between nuclear magnetic moment μ and nuclear angular momentum I)
B= strength of the magnetic field at the nucleus
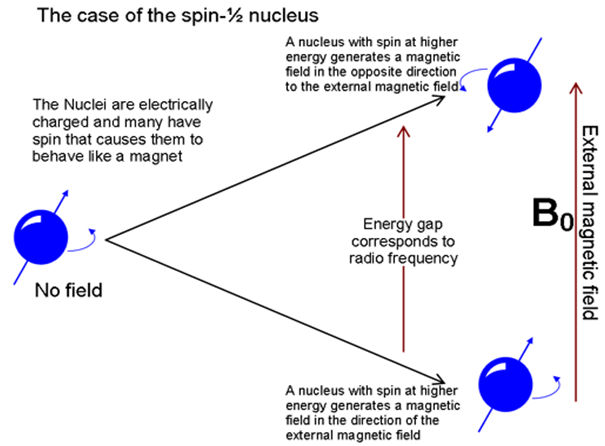
Figure: 3
The precessing proton will absorb energy only if from radiofrequency source only if the precessional frequency is same as the frequency of r.f. beam.
Hence when proton is exposed to external magnetic field the protons will precess , they may not all precess at the same frequency these precessing protons will absorb radio frequency energy of the appropriate frequency and promote proton from lower energy state to higher energy state, this energy absorption is recorded in the form of NMR spectra.
The only nucleus that exhibit NMR phenomenon are those for which spin quantum no (I) is greater than 0…
I is associated with mass no and atomic no.
|
Mass no |
Atomic no |
I |
e.g. |
|
Odd |
Odd/even |
½,3/2,5/2 |
1H1.17O8 |
|
Even |
Even |
0 |
12O6,34S16 |
|
Even |
odd |
1, 2, 3, … |
1H2,7N14 |
All nuclei having I=0 are non magnetic
Other imp magnetic nuclei that have been studied are 11B, 14N, 17O, 19F, 31P
The frequency at which absorption occur can be used for qualitative analysis.
The decrease in intensity of incident radiation owing to absorption during a particular transition is related to no of nuclei in the sample that undergo transition which can be use for quantitative analysis.
Strong magnetic fields are necessary for nmr spectroscopy. The international unit for magnetic flux is the tesla (T). The earth's magnetic field is not constant, but is approximately 10-4 T at ground level. Modern nmr spectrometers use powerful magnets having fields of 1 to 20 T.
NMR instruments typically have a constant Rf and a variable B0.A proton should absorb Rf of 60 MHz in a field of 14,093 Gauss (1.4093 T).Each unique probe nucleus (1H perhaps) will come into resonance at a slightly different - and a very small percentage of - the Rf. All protons come into resonance between 0 and 12/1,000,000 (0 – 12 ppm) of the B0.
* ASPECT OF 1H-NMR SPECTRA:
1. No. of signals present in NMR spectra depend upon no. of type of proton.
2. Intensity of signal depend upon no. of proton under each type.
3. Position of signals tells about chemical environment.
4. Splitting of signal tells about chemical environment.
* Chemical Equivalence.
How many signals in 1H NMR spectrum?
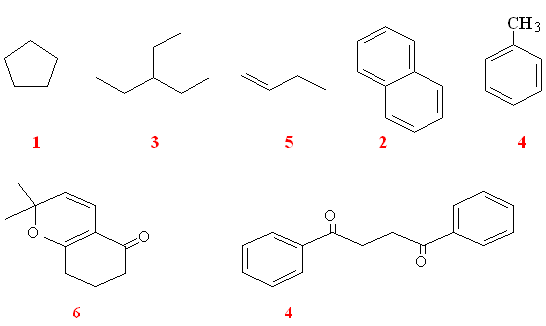
Figure: 4
• Homotopic H’s
Hydrogens are chemically equivalent or homotopic if replacing each one in turn by the same group would lead to an identical compound.
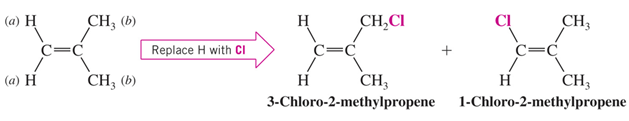
Figure: 5
• Enantiotopic H’s
If replacement of each of two hydrogens by some group leads to enantiomers, those hydrogens are enantiotopic .
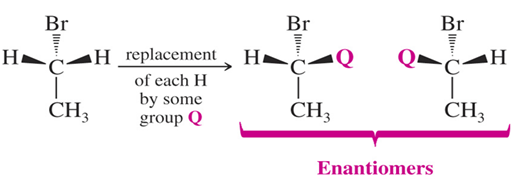
Figure: 6
• Diastereotopic H’s
If replacement of each of two hydrogens by some group leads to diastereomers, the hydrogens are diastereotopic .Diastereotopic hydrogens have different chemical shifts and will give different signals.
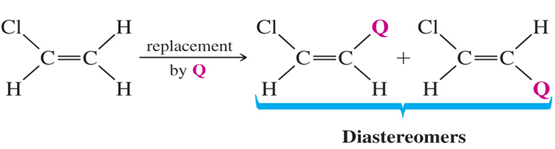
Figure: 7
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
* CHEMICAL SHIFT:
Proton in a molecular are connected other atom – carbon, oxygen, nitrogen, and so on- by covalent bonds. Indeed all the electrons in a molecule, affect the magnetic environment of the protons. Alone, a proton would feel the full strength of the external field, but a proton in an organic molecule respond to both the external field plus any location field within the molecule. An external magnetic field affect the motion of the electron in a molecule, including local field characterized by lines of force that circulate in the opposite direction from the applied field . Thus the net field felt by a proton in a molecule will always be less than applied field, and the proton is said to be a shielded. All of the protons of a molecule are shielded from the applied field by the electron s, but some are less shielded than others. Sometimes the terms “deshielded”, is used to described this decresed shielding of one proton relative to another.
The more shielded a proton is, the greater must be the strength of the applied field in order to achieve resonance and produce a signal. A more shielded proton absorbs rf radiation at higher field strength (up field) compared with one at lower field strength (downfield). Different proton gives signals at different field strengths. The dependence of the resonance position of a nucleus that result from its molecular environment is called its chemical shift. This is where the real power of NMR lies. The chemical shift of various proton in a molecule can be different and are characteristic of particular structural features.
Fortunately for the chemist, all proton resonances do not occur at the same position. The precession frequency (νo) varies because the actual magnetic field B at the nucleus is always less than the external field Bo. The origin of this effect is the "superconducting" circulation of electrons in the molecule, which occurs in such a way that a local magnetic field Be is created, which opposes Bo (Be is proportional to Bo). Thus B = Bo - Be. We therefore say that the nucleus is shielded from the external magnetic field. The extent of shielding is influenced by many structural features within the molecule, hence the name chemical shift. Since the extent of shielding is proportional to the external magnetic field Bo, we use field independent units for chemical shifts: δ values, whose units are ppm. Spin-spin splitting is not dependent on the external field, so we use energy units for coupling constants: Hz, or cycles per second (in mathematical formulas radians per second are the natural frequency units).
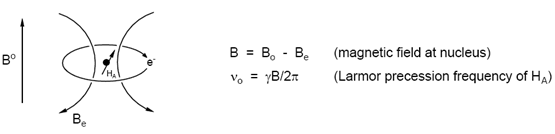
Figure: 8
* The Proton Chemical Shift Scale
One method of solving this problem is to report the location of an nmr signal in a spectrum relative to a reference signal from a standard compound added to the sample. Such a reference standard should be chemically unreactive, and easily removed from the sample after the measurement. Also, it should give a single sharp nmr signal that does not interfere with the resonances normally observed for organic compounds. Tetramethylsilane, (CH3)4Si, usually referred to as TMS, meets all these characteristics, and has become the reference compound of choice for proton and carbon nmr.Experimentally measured proton chemical shifts are referenced to the 1H signal of tetramethylsilane (Me4Si). For NMR studies in aqueous solution, where Me4Si is not sufficiently soluble,thereference signal usually used is DSS (Me3Si-CH2CH2-SO3 -Na+, Tiers, J. Org. Chem. 1961, 26, 2097). For aqueous solution of cationic substrates (e.g., amino acids) where there may be interactions between the anionic reference compound and the substrates, an alternatice reference standard, DSA (Me3Si-CH2CH2-NH3 + CF3CO2 -) has been suggested (Nowick Org. Lett. 2003, 5, 3511). Proton chemical shifts cover a range of over 30 ppm, but the vast majority appear in the region δ 0 10 ppm, where the origin is the chemical shift of tetramethylsilane.1H has a small chemical shift range (15 ppm).
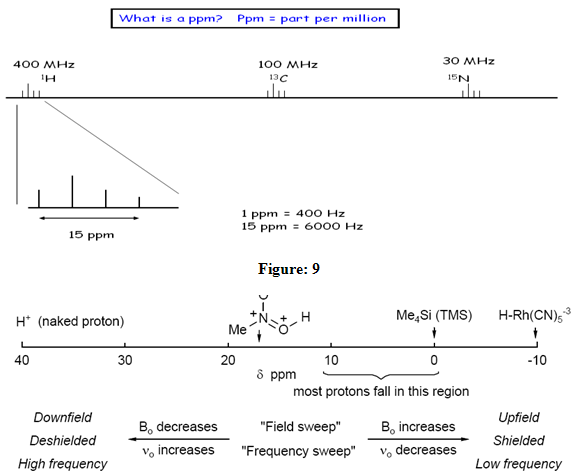
Figure: 10
In the original continuous wave (CW) method of measuring NMR spectra, they were scanned from left to right, increasing the magnetic field. We thus refer to signals on the right as upfield or shielded and signals to the left as downfield or deshielded. Later spectrometers gained the capability of scanning frequency, which then had to decrease from left to right during the scan, hence the "backwards" nature of NMR scales. δ units are defined as follows:
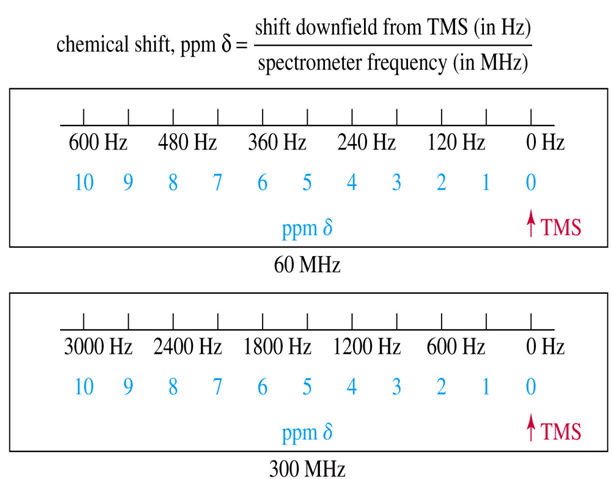
Figure: 11
Chemical shifts of all nuclei should be reported using δ values, with frequency and δ increasing from right to left (many early papers on proton and multinuclear NMR used the opposite convention). Coupling constants are field independent, and should always be specified in Hz.
The chemical shifts of protons on carbon in organic molecules fall in several distinct regions, depending on the nature of adjacent carbon atoms, and the substituents on those carbons. The scale below should be used only as a rough guideline, since there are many examples that fall outside of the indicated ranges. To a first approximation, protons attached to sp3 and sp carbons appear at 0-5 ppm, whereas those on sp2 carbons appear at 5-10 ppm.
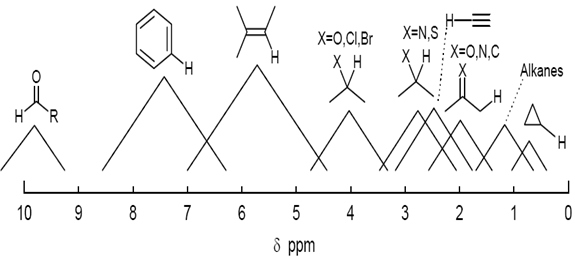
Figure: 12
* Reproducibility of Proton Chemical Shifts:
It is important to understand that the chemical shift of a given proton is not an invariant property of a molecule (like a melting point or boiling point), but will change depending on the molecular environment. The variability is especially large for NH and OH protons (several ppm), but even for CH protons reported shifts vary by a few tenths of a ppm. This is in part due to changes in measurement conditions, but additional variability in chemical shift is present in old NMR data (CW spectra) since spectrometer calibrations and spectrum referencing were not nearly as accurate as they are today. Nevertheless, if conditions are rigorously controlled, very high reproducibility of chemical shifts can be achieved. Databases of precise chemical shifts for many biomolecules have been created which facilitate simultaneous detection by NMR in aqueous solution.
Solvent effects:
The aromatic solvents benzene and pyridine cause shifts as large as 0.5 to 0.8 ppm when compared to less magnetically active solvents like chloroform or acetone. Since the standard solvent for chemical shift parameters like the Curphy-Morrison ones is CCl4 or CDCl3, expect less accurate calculations for spectra taken in aromatic solvents.
Concentration dependence:
Chemical shifts of C-H protons can vary with concentration, especially if intermolecular hydrogen bonding can occur, as for many amines, alcohols and carboxylic acids. The chemical shifts of protons on oxygen (OH) and nitrogen (NH), which are often directly involved in hydrogen bonding are especially strongly dependent (several ppm) on concentration, solvent and temperature.
Temperature dependence:
Chemical shifts can vary with temperature because the chemical shifts of various conformations are different, and the populations of conformations change with temperature (the observed chemical shift is the weighted average of all the shifts of the individual conformations). Temperature can also affect the degree of intermolecular hydrogen bonding or other types of aggregation, and this provides an additional source of shift changes.
Paramagnetic impurities:
(unpaired electrons, transition metals with unpaired spins) can cause very large shifts (tens and hundreds of ppm) as well as large amounts of line broadening. Must avoid these alltogether if you want to get high quality NMR spectra.
* Proton Chemical Shift Effects:
Electronegativit:.
Since the field experienced by the proton defines the energy difference between the two spin states, the frequency and hence the chemical shift, δ/ppm, will change depending on the electron density around the proton. Electronegative groups attached to the C-H system decrease the electron density around the protons, and there is less shielding (i.e. deshielding) so the chemical shift increases or Proton shifts move downfield when electronegative substituents are attached to the same or an adjacent carbon .Alkyl groups behave as if they were weakly electron withdrawing, although this is probably an anisotropy effect.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Table: 1.Proton Chemical Shifts of Methyl Derivatives
|
Compound |
(CH3)4C |
(CH3)3N |
(CH3)2O |
CH3F |
|
δ |
0.9 |
2.1 |
3.2 |
4.1 |
|
Compound |
(CH3)4Si |
(CH3)3P |
(CH3)2S |
CH3Cl |
|
δ |
0.0 |
0.9 |
2.1 |
3.0 |
Table: 2. Proton Chemical Shifts (ppm)
|
Cpd. / Sub. |
X=Cl |
X=Br |
X=I |
X=OR |
X=SR |
|
CH3X |
3.0 |
2.7 |
2.1 |
3.1 |
2.1 |
|
CH2X2 |
5.3 |
5.0 |
3.9 |
4.4 |
3.7 |
|
CHX3 |
7.3 |
6.8 |
4.9 |
5.0 |
|
Lone Pair Interactions:
When lone pairs on nitrogen or oxygen are anti to C-H bonds, the proton is shifted upfield. There is thus a strong conformational dependence of chemical shifts of protons α to heteroatoms. This interaction is one of the reasons that Curphy-Morrison chemical shift calculations work poorly when multiple O or N substituents are attached to one carbon.
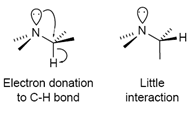
Figure: 13
Magnetic Anisotropy:
Whereas the local circulation of electrons around HA is a shielding effect (i.e., to the right in the NMR spectrum, -δ), there can be both shielding and deshielding effects on HA from electron motion in other parts of the molecule. We refer to such interactions as magnetic anisotropy effects, since they are caused by anisotropic electron circulation (i.e., the electron circulation is stronger in some orientations of the molecule in the magnetic field than in others). The most dramatic examples of anisotropy effects are seen with benzene and other aromatic rings, which cause very large shielding (-δ) effects for protons placed above the ring, and deshielding (+δ) effects for protons to the side of it. These chemical shift effects occur because electron circulation is stronger when the plane of the benzene ring is perpendicular to the magnetic field than when it is parallel to it.
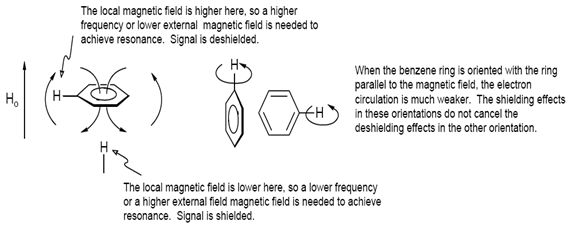
Figure: 14
The consequence of magnetic anisotropy effects is to provide a stereochemical component to the chemical shift of a nucleus: the chemical shift changes depending on the spacial relationship between a proton and nearby functional groups. Such effects can be valuable for making stereochemical assignments. Some proposed magnetic anisotropy shielding/deshielding cones are shown below:
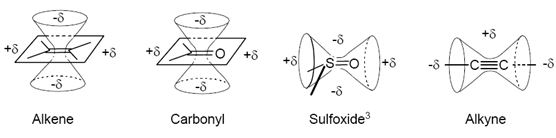
Figure: 15
Solvent Effects:
Chloroform-d (CDCl3) is the most common solvent for nmr measurements, thanks to its good solubilizing character and relative unreactive nature ( except for 1º and 2º-amines). As noted earlier, other deuterium labeled compounds, such as deuterium oxide (D2O), benzene-d6 (C6D6), acetone-d6 (CD3COCD3) and DMSO-d6 (CD3SOCD3) are also available for use as nmr solvents. Because some of these solvents have π-electron functions and/or may serve as hydrogen bonding partners, the chemical shifts of different groups of protons may change depending on the solvent being used. The following table gives a few examples, obtained with dilute solutions at 300 MHz.
|
Table: 3. Some Typical 1H Chemical Shifts (δ values) in Selected Solvents |
||||||||
|
CDCl3 |
C6D6 |
CD3 |
CD3 |
CD3C≡N |
D2O |
||
|
(CH3)3C–O–CH3 |
1.19 |
1.07 |
1.13 |
1.11 |
1.14 |
1.21 |
||
|
(CH3)3C–O–H |
1.26 |
1.05 |
1.18 |
1.11 |
1.16 |
--- |
||
|
C6H5CH3 |
2.36 |
2.11 |
2.32 |
2.30 |
2.33 |
--- |
||
|
(CH3)2C=O |
2.17 |
1.55 |
2.09 |
2.09 |
2.08 |
2.22 |
||
For most of the above resonance signals and solvents the changes are minor, being on the order of ±0.1 ppm. However, two cases result in more extreme changes and these have provided useful applications in structure determination. First, spectra taken in benzene-d6 generally show small upfield shifts of most C–H signals, but in the case of acetone this shift is about five times larger than normal. Further study has shown that carbonyl groups form weak π–π collision complexes with benzene rings, that persist long enough to exert a significant shielding influence on nearby groups. In the case of substituted cyclohexanones, axial α-methyl groups are shifted upfield by 0.2 to 0.3 ppm; whereas equatorial methyls are slightly deshielded (shift downfield by about 0.05 ppm). These changes are all relative to the corresponding chloroform spectra.
The second noteworthy change is seen in the spectrum of tert-butanol in DMSO, where the hydroxyl proton is shifted 2.5 ppm down-field from where it is found in dilute chloroform solution. This is due to strong hydrogen bonding of the alcohol O–H to the sulfoxide oxygen, which not only de-shields the hydroxyl proton, but secures it from very rapid exchange reactions that prevent the display of spin-spin splitting. Similar but weaker hydrogen bonds are formed to the carbonyl oxygen of acetone and the nitrogen of acetonitrile.
Chemical Shift Effects of Phenyl Groups:
The effects of a phenyl substituent are highly dependent on conformation. For example, for styrenes the chemical shift effect of the phenyl is downfield when the phenyl is in the plane of the double bond, but upfield when the rotamer with the phenyl group perpendicular is the more stable one:

Figure: 16
REFERENCES
1. Silverstein RM, Webster FX. Spectrometric identification of organic compounds. 6th ed. Canada: John wiley & sons, Inc, 1998. page no:144-246
2. Kalsi PS. Spectroscopy of organic compound. 6th ed. New Delhi: New age international Pvt. Ltd. Page no:185-370
3. Cary FA. Organic chemistry. 4th ed. New York: McGraw hill. Page no:487-507
4. Reich HJ. NMR Spectroscopy. 1101 University Avenue Department of Chemistry University of Wisconsin Madison, WI 53706, USA. Available from URL: chem.wisc.edu/areas/reich/group/index.htm
5. NMR Spectroscopy URL: 2.chemistry.msu.edu/faculty/reusch/VirtTxtJml/suppmnt1.htm#nom1
6. Medham J, Denney RC, Barnes JD, Thomas M. Vogel’s Text book of qualitative chemical analysis. 6th ed. London: Person education Ltd, 2008. Page no: 599-604.
7. Chatwal GR, Anand SK. Instrumental methods of chemical analysis. 5th ed. Mumbai: Himalaya publishing house. 2005. Page no: 2.185-2.196.
8. Eames J, Peach JM. Stereochemistry at a glance. 1st ed. Australia: Blackwell publishing inc. 2003. Page no: 1-21.
9. Parfitt RT. Nuclear magnetic resonance spectroscopy. In: Backett AH, Stenlake JB, editors. Practical pharmaceutical chemistry. Part-2. 4th ed. New Delhi: CBS publications and distributors. Page no: 408-411, 418-419, 424-431.
10. Willard HH, Merritt LL, Dean JA, Settle FA. Instrumental methods of analysis. 7th ed. India: CBS publishers and distributors. Page no: 422-426, 440-443.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE







