

 About Authors:
About Authors:
Khayyam Shaikh*, Patwekar Shailesh, Santosh Payghan, John Disouza
Department of Pharmaceutics,
Tatyasaheb Kore College of Pharmacy,
Warananagar, Kolhapur, Maharashtra, India 416113.
*ramzanshek0587@gmail.com
ABSTRACT
During the last two decades there has been a remarkable increase in interest in sustained release drug delivery system. This has been due to various factors viz. the extensive cost of developing new drug entities, expiration of existing international patents, discovery of new polymeric materials suitable for prolonging the drug release, and improvement in therapeutic efficacy and safety achieved by this delivery system. A number of design options are available for the preparation of controlled release formulations to modify oral absorption. Formulation approaches are being explored to enhance bioavailability of poorly water-soluble drugs. One such approach that has been shown significantly enhanced absorption of such drugs is to formulate solid dispersionand then formulate its tablets. Solid dispersion technology can be used to improve the in- vitro and in- vivo dissolution properties of slightly water soluble drugs and to control their dissolution rate. In this current study attempts have been made to formulate sustained release tablets of solid dispersions.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1612
INTRODUCTION
The term sustained release has been constantly used to describe a pharmaceutical dosage form formulated to retard the release of a therapeutic agent1.
The term controlled release on the other hand has a meaning that goes beyond the scope of sustained drug action. It also implies predictability and reproducibility in the drug release kinetics, which means that the release of drug ingredients from a controlled release drug delivery system proceeds at a rate profile that is not only predictable kinetically, but also reproducible from one unit to other2.
During the last two decades there has been a remarkable increase in interest in sustained release drug delivery system. This has been due to various factors viz. the extensive cost of developing new drug entities, expiration of existing international patents, discovery of new polymeric materials suitable for prolonging the drug release, and improvement in therapeutic efficacy and safety achieved by these delivery system3. Now-a-days the technology of sustained release has also being applied to veterinary products4.
Figure 1: Drug level versus time.
[adsense:468x15:2204050025]
Advantages of sustained release dosage form5,6,7 : Various advantages of sustained release dosage forms include increased patient compliance due to reduced frequency of drug administration, reduced night time dosing, reduced patient care time, constant supply of drug is provided, decreased local and systemic side effects, reduced gastrointestinal irritation, better drug utilization, reduction in total amount of drug used etc.
Development of controlled release drug delivery systems provide a uniform concentration or amount of drug at absorption site, maintained plasma concentration within a therapeutic range, minimizes the side effects and reduces the frequency of drug administration8. In the last decade, a considerable attention has been focused on the development of novel drug delivery systems because of their obvious advantages such as ease of administration, controlled releases of drug at slower predetermined rate, effectiveness in the treatment at chronic conditions and better patient convenience due to simplified dosing schedule. A number of design options are available for the preparation of controlled release formulations to modify oral absorption. Formulation approaches are being explored to enhance bioavailability of poorly water-soluble drugs. One such approach that has been shown significantly enhanced absorption of such drugs is to formulate solid dispersion9and then formulate its tablets.
Solid dispersion technology: Solid dispersion technology can be used to improve the in- vitro and in- vivo dissolution properties of slightly water soluble drugs and to control their dissolution rate10.Solid dispersion is a product formed by converting a fluid drug carrier combination into solid state11.
Methods of preparation of solid dispersion 11,12: Two methods were used to prepare solid dispersions viz. Melting (fusion) method and Solvent method.
Stability problems of solid dispersions 13: The solid dispersion technique is used for increasing dissolution and absorption rate of poorly soluble drugs. Although the use of solid dispersions has been reported frequently in pharmaceutical literature, very few marketed products rely on solid dispersion strategy. The main reason for this discrepancy is the physical instability (aging effects) of these structures, which are often metastable. Phase separation, crystal growth, or conversion from the amorphous form (metastable form) to crystalline state during storage inevitably results in decreased solubility and dissolution rate.
The proper selection of carrier is often adequate to prevent instability. A carrier that will increase the glass transition temperature (Tg) of the binary system so that molecular mobility becomes extremely low at room temperature, thereby leading to acceptable physical stability. Thus such solid dispersions can be used to formulate sustained release tablets.
Stabilization of lovastatin14: HMG-CoA reeducates inhibitors, stability in an acidic environment is one of a major problems. In order to achieve suitable dosage forms comprising HMG-CoA reductase inhibitors, it is desirable to adequately protect the active ingredient against pH-related destabilization. Stabilization of lovastatin can be achieved by enteric coating of tablets.
Objectives: Lovastatin is a hypolipedimic agent and is insoluble in water as a result, its oral bioavailability is just less than 5% .This necessitates the administration of unnecessarily larger dose of drug. In addition to this the drug has a half-life of 1.1-1.7 hours which requires frequent administration of drug. Thus attempts have been made to formulate solid dispersions and its sustained release tablets with following objectives.
1. To improve solubility and dissolution characteristics of Lovastatin, in order to enhance its oral bioavailability by developing solid dispersions using water soluble carriers.
2. To reduce dose, dosing frequency and dose related side effects by developing sustained release tablets in order to improve patient compliance.
3. To improve the stability of lovastatin in stomach by enteric coating of the tablets.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
MATERIALS AND METHODS
Materials: The chemicals required were Lovastatin(Reddy’s Laboratories Ltd. Hyderabad), Hydroxypropylmethylcellulose (Rexer Pharma Pvt. Ltd. Hyderabad), Crospovidone(Rexer Pharma Pvt. Ltd. Hyderabad), Croscarmellose sodium(Rexer Pharma Pvt. Ltd Hyderabad), Sodium starch glycolate (Rexer Pharma Pvt. Ltd. Hyderabad) and Hydroxypropylmethylcellulose phthalate (Rexer Pharma Pvt. Ltd Hyderabad).
The instruments required were FTIR Spectrophotometer (Model - 8400S, Shimadzu Corporation, Kyoto, Japan), Differential Scanning Calorimeter (METTLER DSC 30S, Mettler Toledo India Pvt. Ltd., Switzerland), Double Beam UV Spectrophotometer (Model No. UV 2401 PC, Shimadzu Corporation, Kyoto, Japan), Digital pH Meter (Model No.335, Systronics, Ahmadabad), Tablet Compression Machine (Type – CMD3 – 16, Cadmach Machinery Pvt. Ltd., Ahmadabad), Tablet Tester (Model No. C – WWTDH 500N, Campbell Electronics, Mumbai), Dissolution test Apparatus (Model No. DA-3, Veego Scientific Devices, Mumbai), Tap Density Tester(Model No. ETD-1020, Electrolab Pvt. Ltd, Goregaon (E), Mumbai), Electronic Weighing balance (Model No. AW-220 and BX - 620S, Shimadzu Corporation, Kyoto, Japan), Heating Humidity chamber(SECOR India, Delhi, India).
Methods:
1. Preparation of solid dispersions11,12: Lovastatin and the superdisintegrants were weighed in different ratio as shown in Table 1 and dissolved in sufficient quantity of acetonitrile, followed by removal of organic solvent by keeping in oven at 40-600C, till constant weight is achieved. The dried dispersions were passed through sieve no.100. The prepared dispersions were stored in glass vials.
Table 1: Composition of solid dispersions
|
Sr.No |
Composition |
Ratio (w/w) |
|
1 |
Lovastatin : Sodium starch glycolate (SSG) (S1) |
1:2 |
|
2 |
Lovastatin : SSG(S2) |
1:4 |
|
3 |
Lovastatin : Croscarmelose Sodium (CCS) (S3) |
1:2 |
|
4 |
Lovastatin : CCS (S4) |
1:4 |
|
5 |
Lovastatin : Crospovidone (CRP) (S5) |
1:2 |
|
6 |
Lovastatin : CRP (S6) |
1:4 |
|
7 |
Lovastatin : SSG : CCS (S7) |
1:2:2 |
|
8 |
Lovastatin : SSG : CRP (S8) |
1:2:2 |
|
9 |
Lovastatin : CCS : CRP (S9) |
1:2:2 |
2. Evaluation of solid dispersions: The formulated solid dispersions were evaluated for different parameters viz. drug content10, solubility15, dissolution studies and their physical properties like bulk density, tapped density, compressibility index and hausner ratio.
3. Formulation of sustained release matrix tablets: After selection of optimized solid dispersion they were further used to prepare sustained release matrix tablets using HPMC polymer with varying ratios as shown in Table 2.
All ingredients used were passed through sieve No.100, and blended in glass mortar uniformly. After sufficient mixing of all components, magnesium stearate was added and further mixed for additional 2-3 minutes. The tablets were compressed using 12 mm concave punch on a single stroke punching machine.
Table 2: Composition of lovastatin matrix tablets
|
Batch code |
Lovastatin Solid dispersions (mg) |
H.P.M.C.K100M (mg) |
Mg stearate(mg) |
Talc (mg) |
Lactose (mg) |
|
K1 |
20 |
100 |
2.5 |
5 |
122.5 |
|
K2 |
20 |
150 |
2.5 |
5 |
172.5 |
|
K3 |
20 |
50 |
2.5 |
5 |
72.5 |
|
K4 |
20 |
60 |
2.5 |
5 |
162.5 |
|
K5 |
20 |
80 |
2.5 |
5 |
142.5 |
|
K6 |
20 |
70 |
2.5 |
5 |
152.5 |
The tablets were compressed using 12 mm concave punch.
4. Evaluation of sustained release tablets:
i) Uniformity of weight16: Twenty tablets were randomly selected from each batch and individually weighed .The average weight was determined.
ii) Hardness: Hardness was measured using Pfizer hardness tester. For each batch three tablets were tested.
iii) Friability17: Mass of tablets equal to 6.5 g was taken for tablets having weight less than 650mg and for tablets of weight more than 650 mg a sample of 10 whole tablets was taken and placed in a Roche friabilator and rotated for 100 times at 25 rpm and tablets were removed dedusted and weighed again. The % friability was measured using the formula
% F = {1- (W / Wo)} x 100
Where,
% F = Friability in %
Wo = Initial weight of the tablet
W = Weight of tablets after test
iv) Thickness18: Three samples were selected randomly from each batch and thickness was measured using Vernier caliper.
v) Drug content16: Twenty tablets were weighed accurately and powdered. Powder equivalent to 20 mg of drug was taken and to that alcohol was added and shaken for 20 min. on a shaker and then diluted with simulated intestinal fluid to get concentration of 50 μg/ml and absorbance was taken.
vi) Dissolution studies: Dissolution was carried out using USP apparatus II taking 900 ml of the medium simulated gastric fluid (without pepsin) pH 1.2 for first two hours followed by simulated intestinal fluid (without pancreatin) pH 6.8 for rest ten hours. The rotational speed of the paddle was set at 100 rpm. 10 mL of aliquots was withdrawn at predetermined time interval for 12 hours and was being replaced by same volume of fresh medium. The sample were analyzed for drug content using double beam UV spectrophotometer at 286 and 325 nm against blank using SGF and SIF respectively. The dissolution was carried out in triplicate for each formulated batch. The cumulative % drug release was calculated using the equation generated from the standard calibration curve.
vii) Swelling studies19,20:
A. Swelling Study (Water uptake study): Swelling of tablet excipients particles involves the absorption of a liquid resulting in an increase in weight and volume. Liquid uptake by the particle may be due to saturation of capillary spaces within the particles or hydration of macromolecule. The liquid enters the particles through pores and bind to large molecule, breaking the hydrogen bond resulting in the swelling of particle. The extent of swelling can be measured in terms % weight gain by the tablet.
B. Erosion study: Poorly soluble drugs are mainly released by erosion process. The mechanism of the erosion can be explained as follows “as the preparation swells it becomes more susceptible to the erosion, and leads to increase in the release rate of insoluble drugs." The extent of erosion can be measured in terms of % weight loss by the tablet.
For each formulation batch, one tablet was weighed and placed in a USP type 2 dissolution apparatus containing 900 ml of 0.1N HCI, with the paddle speed of 55 rpm. After predetermined time interval the tablet was removed from apparatus, blotted to remove excess water, and weighed on analytical balance, the weighted samples were then dried and in oven at 110 0C for 24 hrs., allowed to cool in a desiccator, and finally weighed until constant weight was achieved (final dry weight). The increase in the wet mass represents the medium uptake (swelling index) while decrease in mass represents the extent of erosion (erosional index).
The % weight gain by the tablet was calculated by the formula,
Swelling Index (S.l.) = {(Wt-Wo)/Wo} x 100
Where,
S.I. = swelling index
Wt = weight of tablet at time t
Wo = weight of tablet before immersion
The % erosion of the tablet mass was calculated by the formula.
Erosional Index (E.I.) : 100 - {(Wt X 100)/Wo}
Where,
E.I. = Erosional index
Wt = weight of table at time t (After drying to constant weight)
Wo = weight of tablet before immersion
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
5. Coating of tablets: In the present study, spray coating method was used to coat the tablets. At first, the tablets were rotated into coating pan for nearly 20 minutes before the actual coating process to smoothen the tablet collars (edges). After this, the tablet were removed from the pan and dedusted and then added to the pan again. The coating polymer solution was sprayed with the help of a chemical sprayer after an interval of 3-4 minutes with subsequent drying with the help of a blower. The process was repeated until the whole polymer solution was sprayed on to the tablets.
Table 3: Compositions of coating solution of various proportions of polymer
|
Batch code |
HPMCP (%w/v) |
|
F1 |
10 |
|
F2 |
5 |
|
F3 |
8 |
Further the Dissolution studies of coated tablets were performed and the data was subjected to kinetic studies.For finding out the mechanism of drug release from combination of hydrophilic and hydrophobic polymer matrix tablet, the dissolution data obtained from the above experiments was treated with the different release kinetic equations21,22.
Zero order release equation
Q = -Ko t + Q0 ---------------------------1
First order release equation
Q = Q0e-Kot-------------------------------2
Higuchi’s square root of time equation
Q = KH t1/2 -------------------------------3
Korse-Meyer Peppas equation
F = (Mt / M) = Km tn ---------------------3
Where,
Q = Amount of drug released at time t
Mt = Drug release at time t
M = Total amount of drug in dosage form.
F = Fraction of drug release at time t
Ko = Zero order release rate constant
KH = Higuchi square root of time release rate constant
Km = Constant depend on geometry of dosage form
n = Diffusion exponent indicating the mechanism of drug release
RESULTS
Evaluation of sustained release tablets:
i) Physical characteristics:
Table 4: Characteristics of sustained release Lovastatin matrix tablets
|
Formulation |
Thickness (mm) |
Hardness (Kg/cm2) |
%Friability |
Drug content (%) |
|
K1 |
5.46±0.05 |
5.56±0.10 |
0.21±0.30 |
97.28±1.26 |
|
K2 |
6.43±0.25 |
5.73±0.20 |
0.24±0.37 |
98.64±1.33 |
|
K3 |
7.46±0.05 |
5.8±0.08 |
0.18±0.53 |
99.24±0.84 |
|
K4 |
5.5±1.25 |
5.36±0.09 |
0.21±0.50 |
97.48±0.66 |
|
K5 |
6.43±0.05 |
5.53±0.06 |
0.13±0.51 |
99.51±0.31 |
|
K6 |
5.84±1.21 |
5.86±0.27 |
0.60±0.67 |
98.89±0.54 |
ii) Dissolution studies:
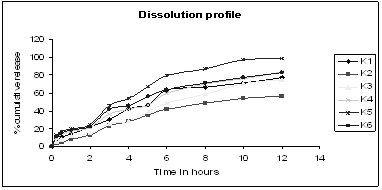
Figure 2: In- vitro release profile of Lovastatin sustained release tablets of batch K1- K7
iii) Swelling studies 69, 70:
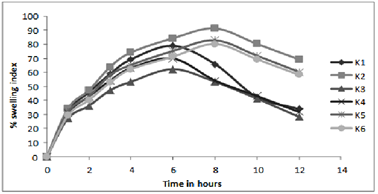
Figure 3:In- vitro profile of swelling index
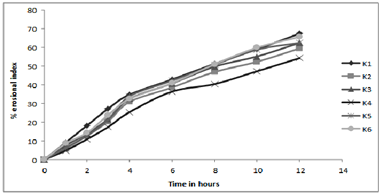
Figure 4:In- vitro profile of erosion index
iv) Dissolution studies of coated tablets: In- vitrorelease profile after HPMCP coating of Lovastatin sustained release tablets of batch F1- F3 was as depicted in figure 5.
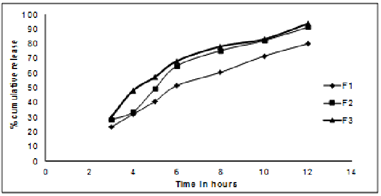
Figure 5: In- vitro release profile after HPMCP coating of Lovastatin sustained release tablets of batch F1- F3
Treatment of dissolution data with different kinetic equations:
Table 5: Kinetic treatment of dissolution data of optimized batch K5
|
S.N. |
Variables |
Zero order |
First order |
Hixon Crowell |
Korsemeyer Pappas |
Higuchi Plot |
|
1 |
r 2 |
0.9860 |
0.9868 |
0.9589 |
0.9269 |
0.9256 |
|
2 |
N |
- |
- |
- |
0.6247 |
- |
|
3 |
K |
13.8609 |
-0.2862 |
-0.0676 |
10.6549 |
26.030 |
Table 6: Kinetic treatment of dissolution data of optimized batch F3
|
S.N. |
Variables |
Zero order |
First order |
Hixon Crowell |
Korsemeyer Pappas |
Higuchi Plot |
|
1 |
r 2 |
0.9559 |
0.9876 |
0.9642 |
0.9298 |
0.9189 |
|
2 |
N |
- |
- |
- |
0.6605 |
- |
|
3 |
K |
14.4173 |
-0.2423 |
-0.0624 |
9.1544 |
24.9804 |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
DISCUSSION
From the standard calibration curve of drug, it was concluded that drug obeys Beer-Lamberts law in concentration range of 0-50mcg/mL.
The dissolution data of plain drug in simulated gastric fluid showed that the release of the drug was less in both the medium and thus it was concluded that Lovastatin is poorly soluble drug and erratically absorbed throughout GI and also possess several dissolution related problem and that might be a reason for its poor bioavailability. Therefore, directly compressed tablet by using superdisintegrants and solid dispersion of drug were prepared by using different polymers. More stable polymers i.e. Crospovidone, Croscarmellose sodium and Sodium starch glycolate were used. The solid dispersions were prepared by solvent evaporation method as it is the easiest to perform and most preferred method.
The drug, solid dispersion and polymer were evaluated for the physical parameters. These physical parameters of solid dispersions and excipients concluded that these were considerably good to formulate the tablet using direct compression technique. The prepared tablets were characterized for physical parameters for each batch. Results reflected that all batches had desirable physical characteristics and had thickness, hardness, content uniformity, and friability values well within permissible range.
In vitro dissolution was carried out for each batch, and dissolution indicated release of Lovastatin which varied according to the quantity of matrix forming polymer. The tablets of batch K1-K6 were prepared using HPMC K100M with varying concentration. As the polymer used had higher the release was 72.22%, 74.97% and 56.21% in 12 hrs respectively. As the release was very less in stipulated time period the use of polymer was further discarded. However tablets of batch K5 sustained the release for 12 hours with cumulative drug release of about 99.08%. From dissolution data it was concluded that drug release rate decreased as the proportion of polymer increased than optimum.
Results of water uptake (swelling) and erosion study cleared that order of swelling observed in these polymers (HPMC) could indicate the rates at which the preparations were able to absorb water and swell. Maximum liquid uptake and swelling of polymer took place in the first 4-8 hours and then gradually decreased due to erosion. Exceptional to above observation batch K4-K6 showed linear increase in swelling as the concentration of the polymer is much greater to dominate the erosion phenomenon.
The observations of swelling and erosion studies of batch K1 to K6 clearly indicated the dual phenomenon i.e., of swelling and erosion, which could be observed visually. This might be due to the presence of poorly soluble Lovastatin with soluble HPMC, which results in the combination of polymer relaxation, which prevails in the first 4-8 hours and erosion, which dominates in the later stages (When erosion of the polymer becomes more intense). These findings were well supported by the Vlachos and co-workers.
The optimized batches of tablets were used for coating with hydroxylpropylmethyl cellulose phthalate of different concentrations of 5%, 8%, 10%. It was observed that tablets coated with HPMCP insoluble in gastric fluid and dissolve rapidly in the upper intestine. Among three batches, F3 (8% HPMCP) showed significant resistance to gastric release of drug and dissolves rapidly in upper intestine.
The optimized batch K5 and F3 were treated with different kinetic equations to interpret the order of release of Lovastatin and the coefficient of determination (r2) was determined. Results indicates that in the selected K5 formulation, the calculated regression coefficients for Zero order, First order, Hixson Crowell, Korsemeyer Pappas, Higuchi Plot models were 0.9860, 0.9868, 0.9589, 0.9269 and 0.9256 respectively. Therefore, the release seemed to fit in theKorsemeyer and Peppas diffusion model and the order in which the drug release was first - order kinetics. In this selected F3 formulation, the calculated regression coefficients for Zero order, First order, Hixson Crowell, Korsemeyer Pappas, Higuchi Plot models were 0.9559, 0.9876, 0.9642, 0.9298 and 0.9189 respectively. Therefore, the release seemed to fit in theKorsemeyer and Peppas diffusion model and the order in which the drug release was first - order kinetics.Further to characterize the release mechanism of Lovastatin from the tablets the dissolution data was subjected to Korsemeyer and Peppas diffusion model.
The values of n (diffusion exponent) were estimated by linear regression of log Mt / M∞ versus log (t). The value of n, for tablet batch K5 was found to be 1.1247 which depicts that the formulation exhibits a non-fickian release behavior, for tablet batch F3 was found to be 1.1605 which depicts that the formulation exhibits a non-fickian release behavior.
REFERENCES
1. Rawlins E.A, 1992. Bentley’s textbook of pharmaceutics, 8th Ed., Bailliere tindall publication, London, pp. 25-29.
2. Jain, N.K., Sharma, S.N., 1998. A Textbook of professional pharmacy, 4th Ed., Vallabh prakashan, New Delhi, pp. 201.
3. United States Pharmacopoeia XXIV NF 19, 2000. United States Pharmacopoeial Convention, Rockville, pp.2236.
4. Udupa, N., Tatawadi, S.V., Gode, K.D., 1985. Pharmaceutical solid dispersions. The Eastern Pharmacist. XXVIII (336), 45-49.
5. Chien, Y.W., 1992. Novel Drug Delivery System, 2nd Ed., Marcel Dekker Inc, New York, pp.1-2.
6. Gudsoorkar, V.R., Rambhau, D., 1994. Sustained release of drugs. The Eastern Pharmacist. XXXVII (443), 87-90.
7. Clarkes analysis of drugs: Pharmaceutical press. Electronic version, 2006 Lovastatin – HMG – CoA reductase inhibitor.
8. Makkiko Fujii, Hideko Okada, Yusuke Shibata, Honami Teramachi, Masuo Kondoh, Yoshiteru Watanabe. Preparation, characterization, and tableting of a solid dispersion of indomethacin with crospovidone. International journal of pharmaceutics 293 (2005) 145-153.
9. Consuelo Souto, Alberto Rodriguez, Silvia Parajes, Ramon Martinez-Pacheco. A comparative study of the utility of two superdisintegrants in microcrystalline cellulose pellets prepered by extrusion-spheronization. European journal of pharmaceutics and Biopharmaceutics 61 (2005) 94-99.
10. Boral, A., Sen, N.L., Ghosh, L.K., Gupta, B.K., 1995. Solid dispersion technology for controlling drug release and absorption. The Eastern Pharmacist. XXXVIII (448), 141-143.
11. Brahmankar, D. M., Jaiswal, S. B., 2003. Textbook of biopharmaceutics and Pharmacokinetics A Treatise, Vallabh Prakashan, New Delhi, pp. 296-302.
12. Leuner, C., Dressman, J., 2000. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm., 50, 47-60.
13. Damian, F., Blaton, N., Kinget, R., Mooter, G.V.D., 2002. Physical stability of solid dispersions of antiviral agent UC-781 with PEG6000, Gelucire 44/14 and PVPK30. Int. J. Pharm., 244, 87-98.
14. Trdna farmacevtska formulacija, Ki vsebuje Lovastatin and Simvastatin, in njena priprva. 09/657,853.
15. Jayaswal, S.B., Subha, P., Gupta, V.K., Vijay Kumar, M., 1994. Studies on dissolution behaviour of sustained release solid dispersions of furosemide. The Eastern Pharmacist. XXXVII (440), 159-161.
16. Indian Pharmacopoeia, 1996. Controller of Publications, Delhi, Vol-I, pp. 236.
17. Sujja-Areevath., Munday, D.L., Cox, P.J., Khan, K.A., 1998. Relationship between swelling, erosion and drug release in hydrophilic natural gum mini-matrix formulations. Eur. J. Pharm. Sci., 6, 207-217.
18. Belgamwar, V.S., Nakhat, P.D., Indurwade, N.H., Avari, J.G., 2002. Development and evaluation of occlusion complexes of griseofulvin with cyclodextrins and their hydroxypropyl derivatives. Indian Drugs. 39(3), 158-160.
19. Sujja-Areevath., Munday, D.L., Cox, P.J., Khan, K.A., 1998. Relationship between swelling, erosion and drug release in hydrophilic natural gum mini-matrix formulations. Eur. J. Pharm. Sci., 6, 207-217.
20. Billa, N., Yuen, K., 2000. Formulation variables affecting drug release from xanthan gum matrices at laboratory scale and pilot scale. AAPS Pharm Sci Tech. 1(4), article 30.
21. Adel M. Aly, M.Semreen, and Mazen K. Qato. Superdisintegrants for Solid dispersion. Pharmaceutical Techlology January 2005, 68-78.
22. Kramer, J., Grady, L. T. and Gajendran, J., 2005. Historical development of dissolution testing, in; Jennifer, D. and Johannes, K., Pharmaceutical dissolution testing, Taylor and Francis, 1-4.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






