

 About Authors:
About Authors:
Jenish.R*, M.Senthil kumar, Dinesh, Ahokkumar, Marshel, Hariharan
Annai veilankanni’s college of pharmacy
Saidapet, Chennai-600015
Tamilnadu
*jenishnathan@gmail.com
ABSTRACT
Alfuzosin is a non-subtype specific alpha(1)-adrenergic blocking agent that exhibits selectivity for alpha(1)-adrenergic receptors in the lower urinary tract. Inhibition of these adrenoreceptors leads to the relaxation of smooth muscle in the bladder neck and prostate, resulting in the improvement in urine flow and a reduction in symptoms in benign prostate hyperplasia. Alfuzosin also inhibits the vasoconstrictor effect of circulating and locally released catecholamines (epinephrine and norepinephrine), resulting in peripheral vasodilation. The alfuzosin of the present investigation is designed to retain in the stomach and deliver the drug alfuzosin for longer periods of time. The developed floating provides increased absorption of the alfuzosin at a rate such that effective plasma levels can be achieved and maintained for a prolonged duration. Formulations 6 displayed drug release considered in 0.1N HCL and Formulation 6 shows better drug release in dissolution profile.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1516
INTRODUCTION
Oral administration is the most convenient and preferred means of any drug delivery to the systematic circulation. Oral controlled release drug delivery have recently been of increasing interest in pharmaceutical field to achieve improved therapeutic advantages, such as ease of dosing administration, patient compliance and flexibility in formulation. This route has high patient acceptability, primarily due to ease of administration. Over the years, oral dosage forms have become increasingly sophisticated with major role being played by 0control release drug delivery system (CRDDS). CRDDS release drug at a predetermined rate, as determined by drug’s pharmacokinetics and desired therapeutic concentration. This help in achieving predictable drug plasma concentration required for therapeutic effect.
The successful functioning of an oral CRDDS is determined by-
1. Physicochemical properties of the drug molecule like, the aquous solubility, intestinal permeability, pH- solubility profile, etc.
2. Pharmacokinetic profile of the drug.
3. The interaction of these properties with the anatomy and physiology of the GI tract.
One such requisite for successful performance of oral CRDDS is that the drug should have the good absorption throughout the GI tract, preferably by passive diffusion.
[adsense:468x15:2204050025]
Drugs that are easily absorbed from gastrointestinal tract (GIT) and have short half- lives are eliminated quickly from the systemic circulation. Frequent dosing of these drugs is required to achieve suitable therapeutic activity. To avoid this limitation, the development of oral sustained-controlled release formulations is an attempt to release the drug slowly into the gastrointestinal tract (GIT) and maintain an effective drug concentration in the systemic circulation for a long time (Ichikawa M et al, 1991). After oral administration, such a drug delivery would be retained in the stomach and release the drug in a controlled manner, so that the drug could be supplied continuously to its absorption sites in the gastrointestinal tract (GIT) (Streubel A et al, 2006). These drug delivery systems suffer from mainly two adversities: the short gastric retention time (GRT) and unpredictable short gastric emptying time (GET), which can result in incomplete drug release from the dosage form in the absorption zone (stomach or upper part of small intestine) leading to diminished efficacy of administered dose (Iannucelli V, et al,1998) . To formulate a site-specific orally administered controlled release dosage form, it is desirable to achieve a prolong gastric residence time by the drug delivery. Prolonged gastric retention improves bioavailability, increases the duration of drug release, reduces drug waste, and improves the drug solubility that are less soluble in a high pH environment (Garg R, et al, 2008). Also prolonged gastric retention time (GRT) in the stomach could be advantageous for local action in the upper part of the small intestine e.g. treatment of peptic ulcer, etc. Gastroretentive drug delivery is an approach to prolong gastric residence time, thereby targeting site-specific drug release in the upper gastrointestinal tract (GIT) for local or systemic effects. Gastroretentive dosage forms can remain in the gastric region for long periods and hence significantly prolong the gastric retention time (GRT) of drugs (Moes AJ et al, 1993). Over the last few decades, several gastroretentive drug delivery approaches being designed and developed, including: high density (sinking) systems that is retained in the bottom of the stomach (Rouge N,et al,.1998), low density (floating) systems that causes buoyancy in gastric fluid (Goole J,et al,. 2007) mucooadhesive systems that causes bioadhesion to stomach mucosa , unfoldable, extendible, or swellable systems which limits emptying of the dosage forms through the pyloric sphincter of stomach, super porous hydrogel systems , magnetic systems etc. (Fujimori J,et al,.1994).
REVIEW OF LITERATURE
Kumaret aldemonstrated works on the gastroretentive dosage forms for prolonging gastric residence time. In the study, the concepts of gastric emptying and absorption windows and current technological developments in gastroretentive drug delivery systems were discussed including their advantages and disadvantages alongwith various evaluation techniques and marketed products for gastroretentive drug delivery. According to the authors, the bioadhesive superporous hydrogel, floating and expanding systems showed the most promising potential for achieving the goal of gastroretention.
El-Kamal et al prepared and evaluated ketoprofen floating oral delivery system. They designed sustained release system for ketoprofen to increase its residence time in the stomach without contact with the mucosa which was achieved through the preparation of floating microparticles by the emulsion-solvent diffusion technique. They used four different ratios of Eudragit S100 with Eudragit RL to form the floating microparticles. It was found that release rates were generally low in 0.1 N HCl especially in presence of high content of Eudragit S100 while in phosphate buffer pH 6.8, high amounts of Eudragit S100 tended to give a higher release rate.
Ali et al formulated hydrodynamically-balanced system for metformin as a single unit-floating capsule.The formulation was optimized on the basis of in vitro buoyancy and in vitro release in simulated fed state gastric fluid. Effect of various release modifiers was studied to ensure the delivery of drug from the HBS capsules over a prolonged period. Capsules prepared with HPMC K4M and ethyl cellulose gave the best in vitro percentage release and were taken as the optimized formulation.
Patel et al developed and optimized a controlled- release multiunit floating system of ranitidine HCl sing compritol, gelucire 50/13 and geliucire 43/01 as lipid carriers. Ranitidine HCl lipid granules were prepared by the melt granulation technique and evaluated for in vitro floatinganddrugrelease. Ethylcellulose, methylcellulose and hyroxypropyl methylcellulose were evaluated as release rate modifiers. They concluded that the hydrophobic lipid Gelucire 43/01 could be considered an effective carrier for design of a multiunit floating drug delivery system for highly water-soluble drugs such as ranitidine HCl.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
MATERIALS AND METHODS
STANDARD GRAPH FOR ALFUZOSIN
The UV scanning of drug sample was carried out using a solution of drug dissolved in methanol solution at concentration of 100 µg/ ml. The λ max was observed at 294nm. The calibration curve of Alfuzosin was obtained by dissolving the drug in methanol solutions and absorbance was measured at 244nm in Methanol solution used as blank. Beer’s law was obeyed the concentration range of 5-25µg in methanol solution.
Method of preparation of pH 6.8 Phosphate buffer solution:
224ml of 0.2M NaOH + 500ml of potassium dihydrogen orthophosphate (KH2PO4) and makeup the volume to one litres.
How to prepare 0.2M NaOH: Dissolve 8 gm of NaOH in 1000ml of distilled water.
How to prepare KH2PO4:Dissolve 27.2 gm of KH2PO4 in 1000ml of distilled water.
EVALUATION PARAMETERS
Precompression Parameters
Bulk Density (Db):
It is the ratio of total mass of powder to the bulk volume of powder. It was measured by pouring the weighed powder into a measuring cylinder and the volume was noted
Tapped Density (DT):
It is the ratio of total mass of powder to the tapped volume of powder. The tapped volume was measured by tapping the powder to constant volume.
Hausner’s ratio:Hausner’s ratio is the ratio of tapped density to bulk density
Invitro dissolution studies: The Invitro dissolution study was carried out in USP dissolution test apparatus type 2 (paddle)
Dissolution Medium: 900ml of simulated gastric fluid
Temperature: 37 ± 0.5 o C RPM: 50
Volume withdrawn & replaced: 5 ml every 60 minutes. Λmax::244 nm.
RESULTS
In the present study, an attempt has been made to formulate and evaluate floating tablets of Alfuzosin by wet granulation method; employing swellable polymers like Hydroxypropylmethycellulose (HPMC K4M), Carbopol940p, aretaken along with other excepients nine formulations are prepared. The formulation is subjected to both pre and post formulation studies.
Hardness and friability: The hardness of the tablet formulations was found to be in the range of 5.1 to 6 kg/cm2(Tables-). Thefriability values were found to be in the range of 0.07 to 0.14 %.( Tables-).
Uniformity of weight: All the prepared tablets of moxifloxacin hydrochloride were evaluated for weight variation. The weight of all the tablets was found to be uniform with low values of standard deviation and within the prescribed IP limits of ±5%.
Uniformity of drug content: The low values of standard deviation indicates uniform drug content within the tablets The percent drug content of all the tablets was found to be in the range of 14.92mg to 15.19mg per tablet (which was within the acceptable limits of ±5%. Tables-)
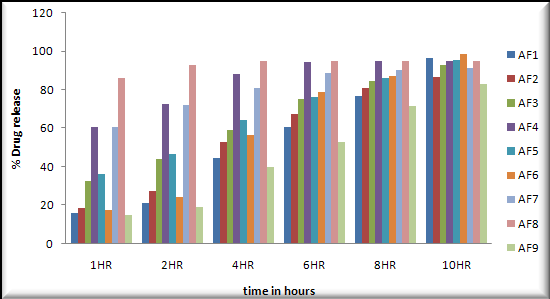
In vitro dissolution study: In vitro dissolution studies were performed in 0.1N HCL on the above promising formulation, namely, formulation 6 .The results are shown in Table- and Table-.
Buoyancy lag Time was observed 04 to 10secnods.
Total floating time (hrs) was observed 10 to 12hours.
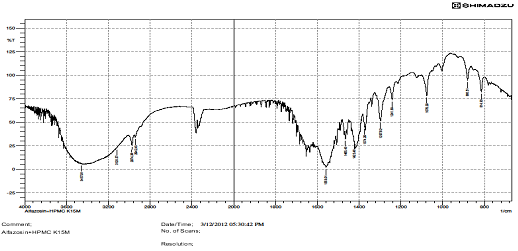
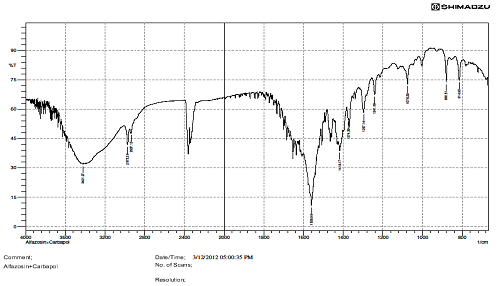
FTIR STUDIES.
There is interaction Find in above IR graphs.IR spectra of drug and formulation along with other excipients are shown in figures.
Short-term stability studies Short-term stability studies on the above promising formulations (at 400C/ 75% RH for 3 months) have shown no significant changes in physical appearance, drug content data of the promising formulation are shown in Tables and figure
TABLES AND GRAPHS
|
Ingredients |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
|
Alfuzosin hydrochloride |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
|
HPMC K15M |
20 |
30 |
- |
- |
20 |
30 |
- |
- |
- |
|
Sodium CMC |
- |
- |
20 |
30 |
20 |
30 |
- |
20 |
30 |
|
Carbopol940 |
- |
- |
- |
- |
- |
- |
20 |
20 |
30 |
|
MCC |
76 |
66 |
76 |
66 |
56 |
46 |
76 |
56 |
46 |
|
Sodium Bicarbonate |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
10 |
|
Magnesium Sterate |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
|
Talc |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
|
Concentration (mcg) |
Absorbance at 244nm |
|
10 |
0.121 |
|
20 |
0.189 |
|
30 |
0.262 |
|
40 |
0.334 |
|
50 |
0.406 |
|
60 |
0.486 |
CONCLUSION
In the present work, floating tablets of Alfuzosin were prepared by Direct Compression method. All the tablets were subjected to weight variation, drug content uniformity, and hardness, and friability, water absorption ratio, wetting time, dissolution, drug excipients interaction and short-term stability studies.
Based on the above study following conclusions can be drawn: Tablets prepared by wet granulation method were found to be good without any chipping, capping and sticking. The hardness of the prepared tablets was found to be in the range of 5.4 to 6 kg/ cm2 The friability values were found to be in the range of 0.07 to 0.14 %. Formulation six showed good results than rest of the nine formulations in pre and post compression studies.
The low values of standard deviation for average weight and drug content of the prepared tablets indicate weight and drug content uniformity within the batches prepared. Formulations 6 displayed drug release considered in 0.1N HCL and Formulation 6 shows better drug release in dissolution profile. Short-term stability studies of promising formulations indicated that there are no significant changes in drug content. IR-spectroscopic studies indicated that there are no drug–excipients interactions.
BIBLIOGRAPHY
1. Kumar R, Philip A. Gastroretentive Dosage forms for prolonging gastric residence time. Int J Pharm Med 2007;21:157-71.
2. El-Kamel AH, Sokar MS, Al Gamal SS, Naggar VF. Preparation and Evaluation of Ketoprofen Floating Oral Delivery System. Int J Pharmaceutics 2001; 220:13-21.
3. Ali J, Arora S, Ahuja A, Babbar AK, Sharma RK, Khar RK, et al. Formulation and development of hydrodynemically balanced system for metformin: In vitro and in vivo evaluation. Eur J Pharmaceutics Biopharm 2007; 67:196-201.
4. Patel DM, Patel NM, Patel VF, Bhatt DA. Floating Granules of RanitidineHydrochloride–Gelucir43/01: Formulation Optimization Using Factorial Design. AAPS Pharm Sci Tech 2007; 8:2.
5. Sahoo SK, Mohapatra S, Dhal SK, Behera BC, Barik BB. Formulation of Floating Microspheres of Ciprofloxacin Hydrochloride by Crosslinking Technique. The Ind Pharmacist 2007; 65:8.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






