

 About Authors:
About Authors:
Nishant Gupta
Department of pharmaceutics,
B.N. College of pharmacy,
Udaipur-313002,
Rajasthan
ABSTRACT:
Paroxetine (PRX) crystals exhibit poor compressibility, poor flowability and its tablets show a tendency to cap. To improve the mechanical strength of tablets several kinds of “Paroxetine for direct compression” are present on the market. Current research demonstrated the best tablet properties with coated paroxetine (mass of tablets, diameter, height and mechanical strength, friability RSD<2%). Furthermore, coated paroxetine in combination with both investigated superdisintegrants such as Vivasol® and Polyplasdone® XL-10 shows faster disintegration time and dissolution rate in comparison to paroxetine for direct compression. Eventually, the major advantages of the formulation with coated paroxetine for industrial production are decrease of friability and superiority in terms of flowability, compressibility, quick disintegration and dissolution. Regarding the results, coating of PRX particles is beneficial for the manufacturing of tablets with immediate release.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1199
INTRODUCTION
Paroxetine (3S,4R )-3-[(1,3-benzodioxol-5-vloxy)methyl]- 4-(4-europhenyl) piperidine is one of the most widely usedis a new generation antidepressant drug. It exerts its antidepressant effect through a selective inhibition for the reuptake of the neurotransmitter serotonin by the presynaptic receptors. PRX is comparable to the tricyclic antidepressants in their clinical effcacy, however, PRX is safer and has greater acceptance by the patients. (Quellet and Percival, 2001). PRX is usually formulated in tablets containing 10 to 40 mg of drug (Martinello et al., 2006). The most appropriate technology for industrial production of tablets is direct compression. In practice, direct compression has been limited mainly to formulation containing small proportions of the active ingredient and appropriate compressibility. The advantages of the direct compression are primary reduced production cost, better product stability and faster dissolution of API when compare to process of granulation (Aulton, 2007). In terms of inadequate compressibility, powder properties can be improved by the addition of binders (Nystrom et al., 1982; Kolter and Flick, 2000). Dry binders can be utilized to improve tablet strength, especially when the interparticulate bonds between the compound crystals are weak (Eichie and Amalime, 2007; Ngwuluka et al., 2010). PRX crystals exhibit poor compression ability, low flowability and its tablets show great tendency to cap. The poor compaction behavior of PRX and its elastic deformation has been related to different PRX polymorphic forms.
Even though they are chemically identical, the different polymorphic forms show different free energies, and different physical properties that can significantly influence product performance. These include differences in solubility and dissolution rate (affecting bioavailability), solid-state stability (affecting potency), deformation characteristics (affecting compressibility), and particle size and shape (affecting powder density and flow properties) (Martinello et al., 2006). There are several methods for modification of the crystalline structure of PRX, in order to manufacture tablets via direct compression.
Examples include spherical crystallization, crystallization from different solvents to produce different crystal habits, incorporation of additives by co-precipitation, development of sintered-like crystals and co-extrusion with isomalt (Di Martino et al., 1996; Fachaux et al., 1995; Garekani et al., 2000; Ndindayino et al., 2001). Untreated PRX particles show massive elastic deformation under pressure which results in axial expansion of tablet in the phase of decompression. As a consequence, tablets often express different types of mechanical strength problems such as capping, chipping, lamination, stress cracking, and sticking as well as picking (Wu et al., 2007).
To improve the mechanical strength of tablets with PRX, several PRXs for direct compression (D.C.) are present in the market. These materials provide sufficient flowability and compactability. PRX for D.C. is modified with different binders, but it still shows optimal dissolution rate, which is necessary for development of tablets with immediate release profile (Fachaux et al., 1995; Kulkarni and Amin, 2008; Okoye et al., 2009). Despite the increasing interest in controlled release drug delivery systems, the most desired forms of tablets are those intended to be swallowed whole. They subsequently disintegrate and release their active ingredient rapidly in the gastrointestinal tract (GIT) (Zhao and Augsburger, 2005a).
Tablets for immediate release often consist of filler, a binder, lubricants and disintegrants (Fukami et al., 2006). In many cases, the disintegration time of solid dosage forms is too long to provide appropriate therapeutic effect. To improve the disintegration time, so-called disintegrants are used. The most accepted mechanisms of their action are wicking, swelling, deformation recovery and particle repulsion. Together, these phenomena create a disintegrating force within the matrix (Zhao and Augsburger, 2005b). In the past, non-modified disintegrants were used to accelerate disintegration, that is, alginates, starches, ambrelite resins, cellulosic materials, pectines and others. Today, a fast working superdisintegrant is chemically modified, typically by crosslinking the organic chains of a polymeric molecules. Three classes of superdisintegrants are commonly used: modified cellulose (croscarmellose sodium - Ac-Di-Sol®, Vivasol®), crosslinked polyvinyl pyrrolidone (Polyplasdone® XL-10) and modified starch (Sodium Starch Glycolate – Primojel®, Explotab®). The basic objective of this study was to produce immediate release tablets containing different types of PRX via direct compression, to compare their properties, disintegration and dissolution profiles. PRX was used as a model drug for immediate release tablets because of its primary indication in treatment of pain where the effect of drug should be rapid. To reach this goal, it is necessary to find a suitable disintegrants having excellent compactability and disintegrating properties. Furthermore, we were focus on establishing differences between PRX powders and to examine how their characteristics influence the tablet manufacture and dissolution profiles. Finally, we wanted to elaborate, whether the coating of PRX particles has a beneficial effect for manufacturing of tablets with immediate release.
MATERIALS AND METHODS
PRX (monoclinic form, Huzhou Konch Pharmaceuticals Co. Ltd.), PRX D.C. (Mallinckrodt inc. St.Louis, USA) and PRX coated (Ethypharm SA, France) were chosen as active ingredients; (Figure 1) Kolidon® VA 64 (Maharashtra, India) and Klucel® - EXF (Maharashtra, India) were used as binders; Polyplasdone® XL-10 (Maharashtra, India) and Vivasol® (Maharashtra, India) were used as superdisintegrants; Avicel® PH 200 (Maharashtra, India) was used as a filler, Mg-stearate (Maharashtra, India) and Aerosil® 200 (Maharashtra, India) were utilized as glidants. Paxil® (Hemopharm concern, Stada, Germany) was chosen as reference tablet.
Powder properties
True, bulk, and tapped density
The true densities of the powder mixtures were determined by helium picnometry (Accupyc 1330, Microneritics, Norcross, Ga., USA). Three samples of each mixture were analyzed, each sample was read three times, and the overall means were calculated. Bulk density measurements were carried out using flat – ground measuring cylinder with a volume of 250 ml. The cylinder was filled with the specified mass of powder mixture and unsettled apparent volume V0 was read to the nearest millilitre. After 10, 250, 500, 1250 taps the corresponding volume was read to the nearest millilitre. The tapped volume was recorded when the difference between the two volumes was smaller than 1 ml For PRX D.C., PRX coated, and tablet powder mixtures, V1250 was used. The tapped density was determined on a tapped volume determination apparatus (Vankel apparatus, Van Kel Technology Group, Edison, NJ, USA).
Apparent density before settling or density of bulk product:
ρ = m/V0 [g/ml] (1)
Apparent density after settling or density of settled product:
ρ = m/V1250 or [g/ml] (2)

Figure 1. Scanning electron micrographs of different paroxetine powders (a - monocrystalline paroxetine; b - paroxetine D.C; c - coated paroxetine).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Flowability
Flowability was defined according to Hausner ratio:
Hausner ratio = (Tapped density) / (Bulkdensity) (3)
Flow of powder was measured using a standard funnel as described in Ph. Eur. VI (European Pharmacopeia, 2008). Into a dry funnel, whose bottom opening has been blocked by suitable means, a test sample was introduced without compacting. After unblocking the bottom opening of the funnel, the time needed for the entire sample to flow out thought the funnel was measured.
Angle of repose
Angle of repose was determined by measuring the height of the cone of powder and calculating the angle of repose - α, from the given equation (Equation 4). The end of a funnel was placed 2 cm above a flat base. The funnel was filled with the powder (the used mass depended on the bulk density of the material, around 2.5 g), therefore after releasing the powder out of the funnel the top of the resulting cone reached the end of the funnel. From the height of the cone (h) and the diameter at the base (d), the angle of repose, (α) was determined.
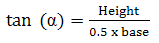 (4)
(4)
Compressibility index
Compressibility index was determined according to Carr’s index:
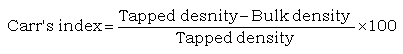 (5)
(5)
Table 1. Direct compression formulation.
|
Ingredients |
Formulation I |
Formulation II |
Formulation III |
Formulation IV |
|
PRX D.C. |
30 |
30 |
- |
- |
|
PRX coated |
- |
- |
40 |
40 |
|
Kolidon VA |
4 |
4 |
2 |
2 |
|
Vivasol© |
- |
5 |
- |
5 |
|
Polyplasdone® XL |
4 |
- |
4 |
- |
|
Avicel PH 200 |
12.5 |
11.5 |
8.64 |
7.64 |
|
Klucel EXF |
2 |
2 |
2 |
2 |
|
Peg 4000 |
1 |
1 |
1 |
1 |
|
Mg-stearate |
1 |
1 |
1 |
1 |
|
Aerosil 200 |
0.5 |
0.5 |
0.5 |
0.5 |
Dry sieve analysis
Particle size distributions of PRX bulk powders, and powder mixtures were determined using a stack of metal sieve plates from the largest to the finest aperture in the following order: 500, 355 (Der Shuenn, Taiwan), 250, 180, 125, 63 and 45 μm (Cole-Palmer, Illinois). Retsch apparatus type AS 200 basic was used for the analysis (F.Kurt Retsch GmbH and Co KG, Germany). The weight of the powders retained on the surface of each sieve plate was divided by the total sample weight to obtain the corresponding weight percentage oversize for each sieve fraction.
Morphology of powder
The scanning electron microscope (Supra 32 VP, Zeiss, Germany) was used to observe the morphology of the PRX powders.
Preparation of tablets
The formulations of PRX tablets are presented in Table 1. Drug and excipients without the lubricants were first mixed for 30 min. Magnesium stearate and Aerosil® 200 are sieved through a sieve (180 μm) and after adding them into the mixture, the mixing was continued for 5 min. Finally, the powder mixture was sieved (sieve 500 μm). Tablets of 55 mg in weight and 4 mm in diameter were prepared by direct compression using a single-punch tableting machine, Kilian SP 300 (Kilian and Co GmbH, Germany). Tablets were compressed with force of 9 kN and compression speed of 25 tbl/min. Finally, the weight and diameter of tablets were measured.
Tablet properties
Uniformity of mass, tablet hardness and friability
The average tablet weight was determined by weighing 20 tablets individually using an analytical balance (European Pharmacopeia, 2008).
Hardness was determined using tablet hardness tester (Vanderkamp VK 200, VanKel Industries, Inc., Edison, NJ, USA). Ten tablets of each formulation were tested. Friability was determined by placing 10 tablets of each formulation in a TAR 10 friabilator (Erweka GmbH, Heusnstamm, Germany) and operating the drum for 4 min and 25 rpm. Friability was determined using the following formula:
Friability = [(Initial weight-Final weight) / Initial weight)] x100 [%] (6)
Measurement of tablet porosity
Tablet porosity (ε) is calculated using Equation 7
ε = 1 - ρa/ρt (7) And
ρ = m/2r2h
where ε is the porosity, ρa is the apparent density, ρt is the true density, m is the mass, r the radius, and h the height of the tablet. The diameter and thickness were determined with a thickness meter (Digitale Schiebelehre, Mister Tool, Walter Werkzeuge Salzburg GmbH, Austria).
Wetting time
The wetting time of tablet was measured by the method described by Bi et al. (1996). The method is as follows. A piece of tissue paper (12 × 10.75 cm) folded twice was placed in a small culture dish, and the time for complete wetting was measured at 25ºC. The wetted tablet was then weighed. Water absorption ratio, R was determined according to the following equation:
R = Wb-Wa
Wa (8)
Where, Wa and Wb are the weight before and after water absorption, respectively.
Disintegration time
Disintegration time was measured with Erweka apparatus Type ZT4 -1, disintegration tester (ERWEKA GmbH, Heusnstamm, Germany). Tests were carried out in 800 ml of distilled water at 37 ± 0.5°C. All tests were run using six tablets of each formulation.
Dissolution study
Dissolution profiles were determined using the paddle method described in USP XXX (United States Pharmacopeia 30th ed., 2007), and the paddle speed of 50 rpm/min (VanKel VK 7000; VanKel industries Inc, Edison, NJ, USA). Dissolution was tested in buffer solutions pH = 1.0 (0.1 N HCl), pH = 4.5 (phosphate buffer), pH = 5.8 (phosphate buffer) and pH = 6.8 (phosphate buffer). The volume of the dissolution medium was 900 ml at 37.0 ± 0.5°C and was prepared according to the Eur. Ph VI. Samples of 10 ml were withdrawn from the dissolution medium at appropriate time intervals and filtered through a membrane filter (pore size 0.45 µm). Each experiment was carried out using six tablets. The samples were appropriately diluted (50×) in a fresh quantity of the dissolution medium. The absorbance was measured by a spectrophotometer (UV-Visible Spectrophotometer 8643, Agilent, France) at 243 nm.
RESULTS
Bulk powder properties
The active ingredients tested in this paper exhibited considerable differences in their powder properties. As shown in Table 2, Hausner ratio, Carr’s index and flow were significantly higher for monocrystalline PRX in comparison to PRX D.C. and coated PRX.
Properties of powder mixtures
Modifications of PRX powder improved the flowability andcompressibility, according to the results presented in Table 3.
Tablet properties
The results of tablet properties are summarized in Table 4. It is apparent that the best tablet characteristics have formulations with coated PRX when compared to formulation with PRX D.C. The maximum official weight variation for tablets heavier than 250 mg is 5%, therefore, all formulations met criteria of the Eur. Ph VI specification (European Pharmacopeia, 2008). Furthermore, all formulations met the specification of Eur. Ph VI (European Pharmacopeia, 2008) for friability of uncoated tablets. Regarding the results, investigated formulations demonstrated a statistically significant decrease of friability in comparison to the reference (p < 0.05) (Table 4).
Table 2. Bulk powder properties.
|
Ingredients |
Mean particle size |
Bulk density (g/ml) |
Flow (s) |
Angle of repose |
Hausner ratio |
Carr’s index |
|
PRX Cryst |
>355 |
0.382 |
59.9 |
∞ |
1.90 |
47.44 |
|
PRX D.C. |
>45 |
0.679 |
35 |
41.63 |
1.39 |
28.12 |
|
PRX coated |
>250 |
0.469 |
30 |
39.52 |
1.14 |
12.36 |
Table 3. Properties of powder mixtures.
|
Ingredients |
Mean particle size (μm) |
Bulk density (g/ml) |
Flow (s) |
Angle of Repose |
Hausner Ratio |
Carr’s index |
|
PRX D.C.+Polyplasdone XL-10 |
>45 |
0.479 |
7.5 |
40.20 |
1.31 |
23.56 |
|
PRX D.C.+Vivasol |
>45 |
0.483 |
8.5 |
41.63 |
1.29 |
22.94 |
|
PRX coated+Polyplasdone XL-10 |
>250 |
0.609 |
8.5 |
39.34 |
1.29 |
18.75 |
|
PRX coated+Vivasol |
>250 |
0.608 |
5.2 |
39.36 |
1.25 |
20.00 |
Table 4. Tablet properties.
|
Ingredients |
Weight (g) |
Porosity (%) |
Crushing strength (N) |
Friability (%) |
Wetting time (s) |
Water/absorption ratio |
|
PRX D.C.+Polyplasdone XL-10 |
0.0559±1.84 |
15.45 |
53.19 |
0.52 |
742 |
0.285 |
|
PRX D.C.+Vivasol |
0.0525±0.43 |
59.43 |
50.46 |
0.51 |
195 |
0.402 |
|
PRX coated + Polyplasdone XL-10 |
0.0512±0.03 |
59.23 |
71.99 |
0.24 |
140 |
0.479 |
|
PRX coated + Vivasol |
0.0562±0.52 |
59.42 |
65.30 |
0.24 |
158 |
0.755 |
|
Reference |
0.0551±0.10 |
- |
61.30 |
0.91 |
178 |
0.897 |
Disintegration time and dissolution profiles
The times required for complete disintegration in water (37ºC, 800 ml) are acceptable according to Eur. Ph VI foruncoated tablets (European Pharmacopeia, 2008) for allinvestigated formulations. Dissolution experimentsperformed on six samples from each formulation aresummarized in Figure 2 (a, b, c, d). According to the results, all formulations met the BCS specification for immediate release dosage forms.


NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
DISCUSSION
The behaviour of the bulk solids is clearly influenced by shape and surface characteristics of the particles integrating a powder (Holgado et al., 1996). The parameters of flowability clearly show that monocrystalline PRX is absolutely inappropriate for direct compression. Angle of repose could not be determined for unmodified PRX because its rheological characteristics were very poor. Prismatic shape of PRX crystals and high surface compression characteristics. In the case of PRX D.C and coated PRX, smaller values of Hausner ratio and Carr’s index theoretically predict better flowability and compressibility. The particle size determination (Table 2) shows that PRX D.C. has the finest particles, which should therefore be able to rearrange in phase of compression and create new surfaces. As a consequence, the tablets will have increased mechanical strength. Furthermore, PRX D.C. has needle shape particles, which increases the specific contact surface area and therefore, compressibility.
The best flow properties were found for coated PRX, which is related to its spherical shape (Figure 1). Particles of coated PRX are very porous and crystals of the active ingredient are implemented inside of the particles. Minimal contact of PRX crystals between coated PRX particles significantly improves flowability and compressibility.
Because of the high content of PRX in the mixture, PRX’s physical characteristics will have an essential influence on the compression properties and elastic relaxation of the tablets. This could be explained with dominant PRX-PRX interactions, which are responsible for more cohesive properties of the mixture and consequently poor flowability. Also, these interactions have an influence on elastic recovery, which has been associated with the storage of elastic energy during compression, as deformation energy under stress, and subsequent release of this energy after removal of axial pressure. On the other hand, spherical structure and protection of PRX crystals inside of coated particles provided compression of higher percentage of active ingredients in comparison to PRX D.C. It was impossible to compress the powder mixture of unmodified PRX with the automatic method on single punch tableting machine because of very high elastic relaxation and capping of the resulting tablets. Furthermore, it was infeasible to make tablets with lactose superdisintegrants (Starlac®, Microcellac®, Ludipress®) and crystalline PRX due to the fact that in these kinds of formulations lactose has limited ability to form strong tablets and it has low dilution potential.
Tablet properties
The mechanical strength of tablets is often defined as the force required fracturing a tablet across its diameter (Martinello et al., 2006). Mechanical strength is directly related to porosity and disintegration time. The packaging process and transportation of the final product requires appropriate tablet strength. Properties of powder mixtures show that PRX D.C. provides greater crushing strength which is in correlation with smaller particle size and higher specific surface area that assures new contact surfaces and bonding between particles. Furthermore, crushing strength of formulations with Polyplasdone® XL- 10 is increased in comparison to formulations containing Vivasol®. The reasons for this are higher compressibility of Polyplasdone® XL-10 (Desai et al., 1994), (unique particle morphology) and the greater content of microcrystalline cellulose - MCC (Avicel PH® 200). Avicel PH® 200 has a nominal mean particle size of 180 _m and because of increased surface area, enables direct compression of mixtures with higher content of PRX than Avicel PH® 101 (Martinello et al., 2006, Rowe et al., 2004). In addition, highly compactable Avicel PH® 200 contributes to greater crushing strength of tablets, which could be explained with the increase in number of mechanical interlocking between MCC particles. Small crushing strength of tablets with coated PRX is associated with particle size, shape and coating of PRX particles. The larger granules with smaller surface area and higher percentage of voids inside of such tablets with consequently weaker inter-particulate bonding requiring the lower crushing strength for diameter fracture (Eichie and Kudehinbu, 2009).
When pharmaceutical powders are compacted into tablets, elastic recovery is responsible for capping, lamination or chipping phenomena. There are different theories to explain capping tendencies, such as entrapment of air, elastic characteristics of materials, radial relaxation of tablets and non-uniform density distribution (Wu et al., 2007).
As mentioned previously, it was not possible to manufacture tablets with unmodified PRX because of the high degree of axial relaxation and tendency for capping. Furthermore, tablets containing modified PRX also showed tendency to cap. Solution for this was usage of Klucell® EXF (hydroxypropylcellulose – HPC) binding agent for direct compression, in concentration of 2% in each formulation. HPC is nonionic, water-soluble cellulose ether with remarkable thermoplastic characteristics, and it has a very high degree of plastic flow. Low molecular weight, low viscosity grades of HPC (EXF) are recommended as immediate release binders. Therefore, when HPC was utilized, it provided toughness, absorbed the compression energy, and finally decreased the ejection force.
Recently, newer classes of natural binders with excellent properties regarding tabletability (stronger interparticulate cohesive bonds) that are also related for manufacturing of tablets with immediate release have been involved (Eichie and Amalime 2007; Ngwuluka et al., 2010; Martins et al., 2007).
Disintegration and dissolution profiles
One of the aims of this paper was also to produce immediate release tablets, which is especially important for treatment of depression. We used PRX, which is classified in Class II, according to the BCS (high permeability, low solubility) (FDA guidelines, 1995). For drugs in Class II, the principal limitation of its oral absorption is its dissolution rate. Because of that, it is necessary to use more than one dissolution medium as a prognostic tool in the assessment of both drug potential for oral absorption and the bioequivalence of its formulation (Dressman et al., 1998). A tablet is considered to be rapidly dissolving when more than 85% of the labeled amount of drug substance (Q + 5%) dissolves within 30 min in a volume of < 900 ml buffer solution using USP Apparatus I or II (United States Pharmacopeia 30th ed., 2007).
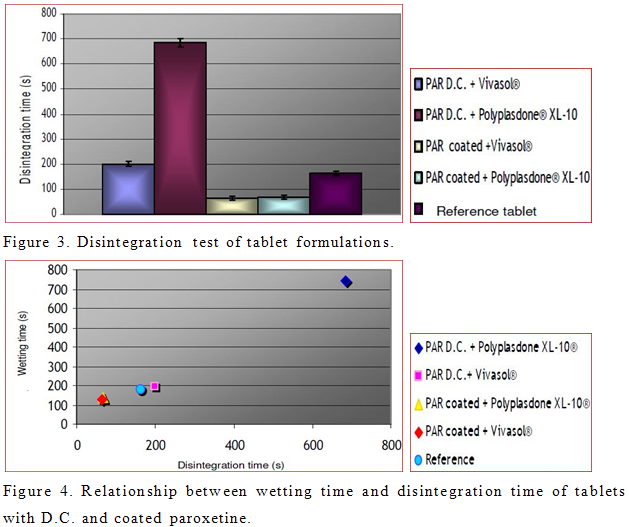
To improve disintegration we used superdisintegrants crosscaramellose sodium (Vivasol®) and cros-spovidone (Polyplasdone® XL-10). Effect of disintegrants and PRX powders on disintegration time of tablets was investigated. Obtained results are shown in Figure 3. However, tablets formulated with coated PRX show faster disintegration time than those prepared with PRX D.C. In the case of coated PRX, present superdisintegrants provided approximately the same disintegration time. On the other hand, different results were obtained in case of tablets with PRX D.C. Tablets formulated from PRX D.C. and Polyplasdone® XL-10 are less porous (Table 4) in comparison to tablets from PRX D.C. and Vivasol®. As a consequence, wicking, which is assumed to be the mechanism of action of Polyplasdone® XL -10 is limited by low porosity of tablets. Possible explanation for decreased disintegration and dissolution of tablets with PRX D.C. and Polyplasdone XL-10® is the very high packaging fraction which obstructs the penetration of fluid into the tablet that would cause the matrix of the tablet to break.
The relationship between the wetting time and disintegration time of tablets is shown in Figure 4.
According to this relationship, as soon as the water penetrates the tablet, it disrupts the interparticulate matrix bonds causing the tablet to fall apart. It has also been reported that crosscaramellose absorbs large amount of water and swells (Table 4). It has been reported that disintegration mechanism of Polyplasdone® XL-10 is wicking, and because of this, tablets containing this superdisintegrant expressed delayed disintegration resulting in filling up the tablet voids (Zhao and Augsburger, 2005b). The dissolution study of the market formulation of PRX (500 mg) demonstrated complete drug release within 30 min. All prepared tablets, except the formulation of PRX D.C. and Polyplasdone® XL-10 showed complete drug release within 30 min. According to results obtained by ANOVA, all formulations show statistically significant differences in dissolution profiles for the first 15 min (data not shown). Also, model independent approach that uses difference factor (f1) and similarity factor (f2) was determined to compare dissolution profiles. Results of fit factors were summarized in Table 5. Generally, f1 values up to 15 (0 to 15) and f2 values greater than 50 (50 to 100) guarantied equivalence of the two dissolution curves and furthermore test and reference similarity (Costa et al., 2003). According to fit factors, formulation with PRX D.C. and Polyplasdone® XL-10 is different in comparison to the reference, while other formulations exhibited similarity in dissolution profiles.
Elaboration of how pH affects the efficiency of the superdisintegrants in terms of dissolution is completely different among scientists. It was reported that, when the superdisintegrant was incorporated into the tablet, dissolution within 15 min was slower in the acidic medium than that in the neutral medium regardless of whether crosspovidone or crosscaramellose was used (Gordon et al., 1993). Also it was pointed out that in a capsule formula (PRX + dibasic calcium phosphate + magnesium stearate) the dissolution was faster in 0.1 N HCl than in deionized water due to the dibasic calcium phosphate excipients (Dahl et al., 1991). Furthermore, Chen et al. (1997) reported that interaction between ingredients in tablets with PRX may lead to different dissolution rates, especially in the acidic medium (Chen et al., 1997). Moreover, Zhao and Augsburger (2005a) demonstrated different dissolution rates from lactose and dicalcium phosphate tablets where croscarmellose was used as superdisintegrant in acidic and neutral media. They also established the same behaviour of Polyplasdone® XL-10 in both media. In our experiment, dissolution data from tablets containing PRX D.C. and Polyplasdone® XL-10 were significantly different in all investigated dissolution media and in all time points when compare to other formulations. This result is in correlation with the small porosity of tablets and longer wetting and disintegration time (Murkesh et al., 2007).
*Significance level (p < 0.05).
|
Fit factors |
pH 1.0; t = 5, 10, 15, 20, 25, 30 |
pH 4.5; t = 5, 10, 15, 20, 25, 30 |
pH 6.8; t = 5, 10, 15, 20, 25, 30 |
|||||||||
|
I/V |
II/V |
III/V |
IV/V |
I/V |
II/V |
III/V |
IV/V |
I/V |
II/V |
III/V |
IV/V |
|
|
f1 |
44.2 |
3.30 |
7.10 |
4.9 |
39.9 |
2,.9 |
9.8 |
8.1 |
42.3 |
7.2 |
10.8 |
10.4 |
|
f2 |
17.3 |
64.7 |
56.9 |
50.9 |
19.7 |
71.3 |
44.0 |
50.5 |
18.7 |
54.0 |
41.1 |
46.3 |
Table 5. Fit Factors, I-Paroxetine D.C. + Polyplasdone XL-10®; II-Paroxetine D.C. + Vivasol®; III -Paroxetine coated + Polyplasdone XL-10®; IV- Paroxetine coated + Vivasol®; V-Reference.
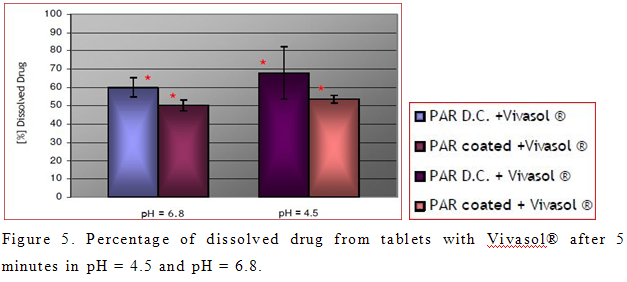
Percentage of dissolved active principle from tablets with PRX D.C. and Polyplasdone® XL-10 did not vary significantly in acidic and neutral media in all time points. This was also confirmed for formulation with coated PRX and Polyplasdone® XL-10, which also verify that behaviour of Polyplasdone® XL-10 is not dependent of pH (Buckton, 1990). Mild agitation force exerted by dissolution paddles (50 rpm) was not strong enough to cause complete breakdown of the larger fragments of tablet that resulted in smaller percentage of dissolved drug in comparison to tablets with Vivasol®. Na Zhao and L. L. Augsburger also confirmed this fact (Zhao and Augsburger, 2005a).
On the other hand, percentage of dissolved active principle from tablets with Vivasol® was greater in acidic medium than in neutral (Figure 5). This result was not expected, because of the decrease swelling capacity of Vivasol® in acidic medium (Zhao and Augsburger, 2005b). Possible explanation for this is the formation of a gel layer in neutral pH, which presents a depot of PRX. However, this result is desired in case of immediate release tablets, which are intended to disintegrate in the stomach, and provide a fast effect. Statistically different percentage of dissolved PRX between tablets with Vivasol® is evident only in the first 5 min in pH = 4.5 and pH = 6.8 (Figure 2b, 2d). It is obvious that dissolution of drug in pH = 6.8 is delayed. This is in correlation with greater swelling and gelling capacity of crosscaramellose in neutral pH (Figure 2d)
Considering the results, best disintegration time and dissolution profile were obtained for coated PRX. Production of tablets with coated PRX is less variable, which is confirmed by appropriate flow of its powder, uniform mass of tablets, improved friability, and disintergration time. Furthermore, presented superdisintegrants provide approximately the same disintergration of prepared tablets. These results confirm positive effect of coating of PRX particles on manufacturing of tablets with immediate release.
REFERENCES
• Aulton EA (2007). Aulton’s Pharmaceutics-The design and manufacture of medicines. In Churchill Livingstone ed. 3rd edition, p.443. Buckton G (1990). The role of compensation analysis in the study of wettability, solubility, disintegration, and dissolution. Int. J. Pharm., 66: 175–182.
• Bi Y, Sunada H, Yonezawa Y, Danjo K, Otsuka A, Iida K (1996). Preparation and evaluation of a compressed tablet rapidly disintegrating in the oral cavity. Chem. Pharm. Bull., 44(11): 2121- 2117.
• Chen C, Lin Y, Cho S, Yen S, Wu H (1997). Investigation of the dissolution difference between acidic and neutral media of paroxetine tablets containing a super disintegrant and a soluble excipient. Chem. Pharm. Bull., 45(3): 509-512.
• Costa FO, Sousa JJS, Pais CC, Formosinho SJ (2003). Comparison of dissolution profiles of ibuprofen pellets. J. Contr. Rel., 89: 199-212.
• Dahl TC, Sui IT, Yum A (1991). The Influence of Disintegrant Level and Capsule Size on Dissolution of Hard Gelatin Capsules Stored in High Humidity Conditions. Drug. Dev. Ind. Pharm., 17(7): 1001-1016.
• Desai DS, Rubitski BA, Bergum JS, Varia SA (1994). Effects of different types of lactose and disintegrant on dissolution stability of hydrochlorothiazide capsule formulation. Int. J. Pharm., 110: 257- 265.
• Di Martino P, Guyot-Hermann A-M, Conflant P, Drache M, Guyot J-C (1996). A new pure paroxetine for direct compression: the orthorhombic form. Int. J. Pharm., 128:1-8.
• Dressman JB, Amidon GL, Reppas C, Shah VP (1998). Dissolution testing as a prognostic tool for oral drug absorption: Immediate release dosage forms. J. Pharm. Res., 15 (1): 11-22.
• European Pharmacopeia, (2008). 6th Edition, Council of Europe, Strasbourg.
• Eichie FE, Amalime AE (2007). Evaluation of the binder effects of the gum mucilages of Cissus populnea and Acassia senegal on the mechanical properties of paroxetine tablets. Afr. J. Biotech. 6(19): 2208-2211.
• Eichie FE, Kudehinbu AO (2009). Effect of particle size of granules on some mechanical properties of paroxetine tablets. Afr. J. Biotech., 8(21): 5913-5916.
• Fachaux JM, Guyot-Hermann AM, Guyot JC, Conflant P, Drache M, Veseler S, Boistelle R (1995). Pure paroxetine for direct compression Part I. Development of sintered-like crystals of paroxetine. Powder. Techol., 82: 123-128.
• Food and Drug Administration. Guidance for industry (1995). Immediate release solid oral dosage forms, Waiver of in vivo bioavailability and bioequivalence studies for immediate release solid oral dosage forms based on biopharmaceutics classification systems. CDER, USA.
• Fukami J, Yonemoshi E, Youshihashi Y, Terada K (2006). Evaluation of rapidly disintegrating tablets containing glycine and carboxymethylcellulose. Int. J. Pharm., 310: 101-109.
• Garekani HA, Ford JL, Rubinstein MH, Rajabi-Siahboomi AR (2000). Highly compressible paroxetine - II. Compression properties. Int. J. Pharm., 208: 101-110.
• Gordon MS, Ruddararaju VS, Dani K, Chowhan ZT (1993). The effect of aging on the dissolution of wet granulated tablets containing super disintegrants. Int. J. Pharm., 97: 119-131.
• Kolter K, Flick D (2000). Structure and dry binding activity of different polymers, including Kollidon® VA 64. Drug. Dev. Ind. Pharm., 26(11): 1159-1165.
• Kulkarni RB, Amin PD (2008). Masking of unpleasant gustatory sensation by cross-linking of dehydrated paroxetine alginate pellets produced by extrusion-spheronization. Drug. Dev. Ind. Pharm., 34: 199-205.
• Martinello T, Kaneko TM, Robles Velasco MV, Santos Taqueda ME, Consiglieri VO (2006). Optimization of poorly compactable drug tablets manufactured by direct compression using the mixture experimental design. Int. J. Pharm., 322: 87-95.
• Martins E, Christiana I, Olobayo K (2007). Effect of Grewia gum on the mechanical properties of paroxetine tablet formulations. Afr. J. Pharm. Pharmacol., 2: 001-006.
• Murkesh CG, Rajesh KP, Brahmbhatt BK, Shah AR (2007). Improving and tablet characteristics and dissolution profile of ibuprofen by using a novel coprocessed superdisintegrant: A technical note. AAPS Pharm. Sci. Tech., 8: 13.
• Ndindayino F, Henrist D, Kiekens F, Van den Mooter G, Vervaet C, Remon JP (2002). Direct compression properties of melt extruded isomalt. Int. J. Pharm., 235: 149-157.
• Ngwuluka NC, Idiakhoa BA, Nep EI, Ogaji I, Okafor IS (2010). Formulation and evaluation of paroxetine tablets manufactured using the dried fruit of Phoenix dactylifera Linn as an excipients. Res. Pharm. Biotech., 2(3): 25-32.
• Nystrom C, Mazur J, Sjögren J (1982). Studies on direct compression of tablets II. The influence of the particle size of a dry binder on the mechanical strength of tablets. Int. J. Pharm., 10: 209-218.
• Rowe RC, Sheskey PJ, Owen SC (2004). Handbook of pharmaceutical excipients. 108-110, Pharmaceutical Press.
• Okoye EI, Onyekweli AO, Ohwoavworhua FO, Kunie OO (2009). Comparative study of some mechanical and release properties of paroxetine tablets formulated with cashew tree gum, povidone and gelatin as binders. Afr. J. Biotech., 8(16): 3970-3973.
• Ouellet M, Percival MD (2001). Mechanism of paroxetine inhibition of cyclooxygenase isoforms. Arc. Biochem. Biophys., 387: 273-280.
• United States Pharmacopeia (2007). 30th Edition, National Formulary 25th ed., United States Pharmacopeia Convection, Rockville, MD, USA.
• Wu CY, Hancock BC, Mills A, Bentham AC, Best SM, Elliott JA (2007). Numerical and experimental investigation of capping mechanisms during pharmaceutical tablet compaction. Powder. Technol., 181(2): 121-129.
• Zhao N, Augsburger LL (2005a). The influence of swelling capacity of superdisintegrants in different pH media on the dissolution of hydrochlorothiazide form directly compressed tablets. AAPS Pharm. Sci. Tech., 6(1): 19.
• Zhao N, Augsburger LL (2005b). Functionally comparison of 3 classes of superdisintegrants in promoting aspirin tablet disintegration and dissolution. AAPS Pharm. Sci. Tech., 6(4): 79.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






