

{ DOWNLOAD AS PDF }
 ABOUT AUTHORS
ABOUT AUTHORS
Amiya Kumar Ghosh*1,Saptarshi Samajdar2, Shiladitya Palit3, Rupchand Pandit4
1Department of Pharmaceutical Technology, UTKAL UNIVERSITY, Bhubaneswar- 751004,Odisha, India.
2Centre for Pharmaceutical Sciences and Natural Products, CENTRAL UNIVERSITY OF PUNJAB, Bathinda-151001, Punjab, India
3Department of Pharmaceutics, IITBHU, Varanasi, India
4Department of Pharmaceutical Science & Technology, Birla Institute of Technology, Mesra Ranchi-835215, Jharkhand, India
*amiyaghosh94@gmail.com
ABSTRACT
Tuberculosis (TB) has been declared as a global health emergency by the World Health Organization(WHO). This has been mainly due to the emergence of multiple drug resistant strains and the synergy between tubercle bacilli and the human immunodeficiency virus (HIV). Mycobacterium tuberculosis (Mtb) is a pathogenic bacteria species in the genus Mycobacterium and the causative agent of most cases of tuberculosis. Tuberculosis (TB) is the leading cause of death in the world from a bacterial infectious disease. This antibiotic resistance strain lead to development of the new antibiotics or drug molecules which can kill or suppress the growth of Mycobacterium tuberculosis.The need for new antiTB is persistent due to the emergence of drug resistant Mycobacterium tuberculosis. Here we aimto identify new drug targets in Mycobacterium tuberculosis by phylogenomics among the Mycobacterium tuberculosisandcomparative genomics to Homo sapiens. The proposed target discovery pipeline is largely independent of experimental data and based on the assumption that Mycobacterium tuberculosis proteins are likely to be essential if (i) there are no similar proteins in the same proteome and (ii) they are highly conserved across the Mycobacterium tuberculosisof mammals. We have performed an in silicocomparative analysis of metabolic pathways of the host Homo sapiens and the pathogen Mycobacterium tuberculosis (H37Rv). Novel efforts in developing drugs that target the intracellular metabolismof M. tuberculosis often focus on metabolic pathways that are specific to M. tuberculosis. We have identified five unique pathwaysfor Mycobacterium tuberculosis having a number of 60 enzymes, which are nonhomologous to Homo sapiens protein sequences,and among them there were 55 enzymes, which are nonhomologous to Homo sapiens protein sequences.These enzymes were alsofound to be essential for survival of theMycobacteriumtuberculosisaccording to the DEG database. Further, the functional analysis using Uniprot showed involvement of all the unique enzymes in the different cellular components.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-2502
|
PharmaTutor (Print-ISSN: 2394 - 6679; e-ISSN: 2347 - 7881) Volume 5, Issue 7 Received On: 16/03/2017; Accepted On: 22/03/2017; Published On: 01/07/2017 How to cite this article: Ghosh AK, Samajdar S, Palit S, Pandit R;Drug targets identification of Mycobacterium tuberculosis by metabolic pathway analysis: insilico process; PharmaTutor; 2017; 5(7); 43-53 |
INTRODUCTION
Mycobacterium tuberculosis is reputed to have the highest annual global mortality among all of the pathogens[1]. The rise in tuberculosis (TB) incidence over the last two decades is partly due to TB deathsin HIV-infected patients and partly due to the emergence of multidrug resistant strains of the bacteria. This rapid increase in the disease has led to potential funding arrangements aimed at a large efforttowards stopping this disease before it becomes a global epidemic. Due to its slow growth and highvirulence it is extremely difficult to work with the TB bacterium. However, rapidly evolvingmycobacterium genomics with complete genome sequences known along with powerful bioinformaticsapproaches, one can realize better therapeutics and prophylactics in the near future[2]. A comparativegenomic analysis of these species has a potential to reveal the genetic basis of disease phenotypes, whichwill be invaluable for the development of much needed drugs and newer vaccines.Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), remains a major health threat. Each year, 8 million new TB cases appear and 2 million individuals die of TB [3]. Further, about half a million new multidrugresistant TB cases are estimated to occur every year [4]. The existing drugs, although of immense value in controlling the disease to the extent that is being done today, have several shortcomings, the most important of them being the emergence of drug resistance rendering even the frontline drugs inactive. In addition, drugs such as rifampicin have high levels of adverse effects making them prone to patient incompliance. Another important problem withmost of the existing antimycobacterials is their inability to act upon latent forms of the bacillus. In addition to these problems, the vicious interactions between the HIV (human immunodeficiency virus) and TB have led to further challenges for antitubercular drug discovery [5]. One of the classical threats of the tuberculosis epidemic has been the MDR-TB. Use, and often abuseor misuse, of antimicrobial agents has encouraged the evolution of bacteria toward resistance, resultingoften in therapeutic failure. There are evidences that bacteria have the ability to adapt to this deficit andrecover fitness on serial passage[1]. Resistance to antituberculosis drugs has been a problem since theera of chemotherapy began. After dramatic outbreaks of MDR-TB in the early 1990s, resistance becamerecognized as a global problem. MDR-TB now threatens the inhabitants of countries in Europe, Asia,Africa, and the Americas[2]. An understanding of the molecular basis of drug resistance may contribute tothe development of new drugs[6]. Management of MDR-TB relies on prompt recognition and treatment with at least 3 drugs to which an isolate is susceptible. The roles of drug containing environments, and the Immunological status of the host and bacterial molecular mechanisms of development of drug resistance to M. tuberculosis have been examined and results are helpful in implementation of modified drug regimens in tuberculosis control programmes. Multidrug resistant strains of M. tuberculosis seriously threaten tuberculosis control and prevention efforts. Molecular studies of the mechanism of actionof antitubercular drugs have elucidated the genetic basis of drug resistance. Drug resistance in M.tuberculosis has been primarily attributed to the mutations in the drug target genes, however, thepresence of efflux pumps in clinical MDR isolates cannot be ruled out[7]. These mutations lead either toan altered target or to a change in titration of the drug. A diverse array of strategies is already available toassist in rapid detection of drug resistance-associated gene mutations. In spite of remarkable advances inthis area, much remains to be learned about the molecular genetic basis of drug resistance in M.tuberculosis. During the last decade, there has been a marked increase in the number and gravity oftuberculosis cases both in developing countries andin industrialized nations. One of the more insidiousconsequences of this resurgence has been the recentemergence of nosocomial transmission of multi drugresistant strains of M. tuberculosis, thus creatinguntreatable forms of the disease and these strains maybecome widespread. That the various clinical isolatesof M. tuberculosis are geographically partitioned atthe global level. Ahmed et al5,6 has provided evidenceto the concept of geographic genomics[8]. Recently, genome-scale metabolic network reconstructionsfor different organisms have enabled systematic analysesof metabolic functions and predictions of metabolismrelatedphenotypes. By collecting all possible biochemicalreactions for specific organisms, different groups have reconstructed metabolic networks for bacteria, for example,Escherichia coli, Helicobacter pylori, and Chromohalobactersalexigens, eukaryotic microorganisms, mice, and even humans [9–11]. The website of the Systems BiologyResearch Group at the University of California, San Diego(http://gcrg.ucsd.edu/), provides a continuously updated listof genome-scale metabolic network reconstructions. Analysisof metabolic networks can provide insights into anorganism’s ability to grow under specific conditions. Forexample, given a specific set of nutrient conditions, fluxbalance analysis (FBA) ofmetabolic networks can accuratelypredict microbial cellular growth rates. In a recent work, agroup of researchers used an approximate representation of in-host nutrient availability inferred from the literature tosimulate the in-host metabolism of Salmonella typhimurium[12]. Moreover, metabolic network analyses can then be usedto identify organism-specific essential genes by predicting theattenuation of microbial growth of specific deletion mutants[13–15].The computational approach has been used to investigatenovel drug targets in other pathogenic organismssuch as Pseudomonas aeruginosa and in Helicobacter pylori[10, 16].As most currently known, antibacterials are essentiallyinhibitors of certain bacterial enzymes; all enzymes specificto bacteria can be considered as potential drug targets [17].In this study, we have adopted a strategy for comparativemetabolic pathway analysis to find out some potential targetsagainst M. tuberculosis (H37Rv). Only those enzymes which show unique properties than the host were selected as thetarget.Metabolic genes that are essential for pathogen growthbut are not present in humans constitute actual and potential drug targets.
Mycobacterium tuberculosis:
Fig 1. Pic depicting microscopic structure ofM.tuberculosis
Scientific classification:
Domain:Bacteria
Phylum:Actinobacteria
Class:Actinobacteria
Order:Actinomycetales
Family: Mycobacteriaceae
Genus:Mycobacterium
Species:tuberculosis
Other bacteria are commonly identified with a microscope by staining them with Gram stain. However, the mycolic acid in the cell wall of M. tuberculosis does not absorb the stain. Instead, acid-fast stains such as Ziehl-Neelsen stain, or fluorescent stains such as auramine are used. Cells are curved rod-shaped and are often seen wrapped together, due to the presence of fatty acids in the cell wall that stick together. This appearance is referred to as cording, like strands of cord that make up a rope. M. tuberculosis is characterized in tissue by caseating granulomas containing Langhans giant cells, which have a "horseshoe" pattern of nuclei.
Objective:
Here we aim to identify new drug targets in Mycobacterium tuberculosis by phylogenomics among the Mycobacterium tuberculosis and comparative genomics to Homo sapiens.The proposed target discovery pipeline is largely independent of experimental data and based on the assumption that Mycobacterium tuberculosis proteins are likely to be essential if (i) there are no similar proteins in the same proteome and (ii) they are highly conserved across the Mycobacterium tuberculosisof mammals. We have performed an in silico comparative analysis of metabolic pathways of the host Homo sapiens and the pathogen Mycobacterium tuberculosis (H37Rv).
Materials and Methods
KEGG (Kyoto Encyclopedia of Gene and Genome) (http://www.genome.jp/pathways.html) [18] pathway database was used as a source ofmetabolic pathway information.Metabolic pathway identification numbers of the host H. sapiens and the pathogen M. tuberculosis (H37Rv) were extracted from the KEGG database. Pathways which do not appear in the host but are present in the pathogen according to KEGG database have been identified as pathways unique to M. tuberculosis as in comparison to the host H. sapiens. Enzymes in these unique pathways as well as enzymes involved in other metabolic pathways under carbohydrate metabolism, energy metabolism, lipid metabolism, nucleotide metabolism, amino acid metabolism, metabolism of other amino acids, and glycan biosynthesis were identified from the KEGG database. The corresponding protein sequences of enzymes involved in unique pathways were identified and their protein sequences were retrieved in FASTA format from KEGG database. The unique enzymes were further analyzed for essentiality to pathogen by DEG (Database of Essential Genes) database (http://tubic.tju.edu.cn/deg/) [19], and considered cutoffscore was >100 to enhance the specificity of enzyme in M.tuberculosis.The obtained targets genes were further analyzed byUniProt (Universal Protein Resource) (http://www.uniprot.org/) database to find out their functions. This is requiredto find out the surface membrane proteins which could be probable vaccine targets.

Fig. 2 : metabolic pathway of Mycobacterium tuberculosis H37Rv
Fig 3. metabolic pathway of Homo sapiens (human) Results and Discussion
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Identification of Unique Pathways and Potential DrugTargets:Tuberculosis (TB) is a major cause of illness anddeath worldwide, especially in Asia and Africa. Globally, 9.2million new cases and 1.7 million deaths from TB occurredin 2006, of which 0.7 million cases and 0.2 million deathswere in HIV-positive people [2]. The existing drugs haveseveral shortcomings, the most important of them being theemergence of drug resistance.No newanti-Mtb drugs have been developed forwell over20 years. In view of the increasing development of resistanceto the current leading anti-Mtb drugs, novel strategies aredesperately needed to avert the “global catastrophe” forecastby the WHO (World Health Organization). Therefore, computational approach for drug targets identification, specificallyfor Mtb, can produce a list of reliable targets veryrapidly.These methods have the advantage of speed and lowcost and, even more importantly, provide a systems view ofthe whole microbe at a time. Since it is generally believedthat the genomes of bacteria contain genes both with andwithout homologues to the human host.Using computationalapproach for target identification it is very quick to produce a desirable list.In the present study, 5 unique pathways, C5-brancheddibasic acidmetabolism, carbon fixation pathways in prokaryotes,methanemetabolism, lipopolysaccharide biosynthesis, and peptidoglycan biosynthesis with 60 new nonhomologoustargets were identified through in silico comparativemetabolic pathway analysis of Homo sapiens and M. tuberculosisH37Rv using KEGG database. Pathways which are notpresent in the Homo sapiens but present in the Mycobacteriumare designated as unique pathways. Design and targetinginhibitors against these nonhomologous sequences couldbe the better approach for generation of new drugs. Thus total 5 unique metabolic pathways have been taken in M.tuberculosis (Table 1).
|
S. no. |
Pathway name |
Human |
Mycobacterium tuberculosis H37Rv |
|---|---|---|---|
|
1 |
Carbohydrate Metabolism |
|
|
|
1.1 |
C5-Branched dibasic acid metabolism |
Absent |
Present |
|
2. |
Energy Metabolism |
|
|
|
2.1 |
Photosynthesis |
Absent |
Absent |
|
2.2 |
Carbon fixation pathways in prokaryotes |
Absent |
Present |
|
2.3 |
Methane metabolism |
Absent |
Present |
|
3. |
Lipid Metabolism |
|
|
|
3.1 |
Fatty acid elongation in mitochondria |
Present |
Absent |
|
3.2 |
Sphingolipid metabolism |
Present |
Absent |
|
3.3 |
Arachidonic acid metabolism |
Present |
Absent |
|
4. |
Nucleotide Metabolism |
All Present |
All Present |
|
5. |
Amino Acid Metabolism |
All Present |
All Present |
|
6. |
Metabolism of Other Amino Acids |
All Present |
All Present |
|
6.1 |
Phosphonate and phosphinate metabolism |
Absent |
Absent |
|
7. |
Glycan Biosynthesis and Metabolism |
|
|
|
7.1 |
N-Glycan biosynthesis |
Present |
Absent |
|
7.2 |
Various types of N-glycan biosynthesis |
|
Absent |
|
7.3 |
Mucin type O-Glycan biosynthesis |
Present |
Absent |
|
7.4 |
Other types of O-glycan biosynthesis |
Present |
Absent |
|
7.5 |
Glycosaminoglycan biosynthesis—chondroitin sulfate |
Present |
Absent |
|
7.6 |
Glycosaminoglycan biosynthesis—heparan sulfate |
Present |
Absent |
|
7.7 |
Glycosaminoglycan biosynthesis—keratan sulfate |
Present |
Absent |
|
7.8 |
Glycosaminoglycan degradation |
Present |
Absent |
|
7.9 |
Glycosylphosphatidylinositol (GPI)-anchor biosynthesis |
Present |
Absent |
|
7.10 |
Glycosphingolipid biosynthesis—lacto and neolacto series |
Present |
Absent |
|
7.11 |
Glycosphingolipid biosynthesis—globo series |
Present |
Absent |
|
7.12 |
Glycosphingolipid biosynthesis—ganglio series |
Present |
Absent |
|
7.13 |
Lipopolysaccharide biosynthesis |
Absent |
Present |
|
7.14 |
Peptidoglycan biosynthesis |
Absent |
Present |
|
7.15 |
Other Glycan degradation |
Present |
Absent |
Table 1:comparison of metabolic pathway present in M. tuberculosis and H. sapiens
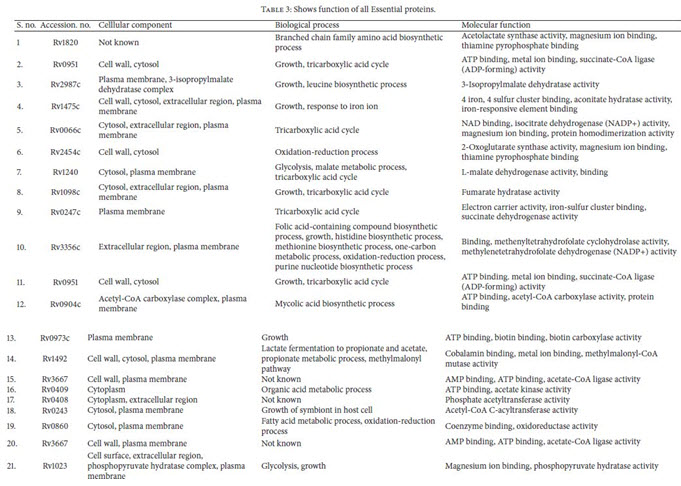
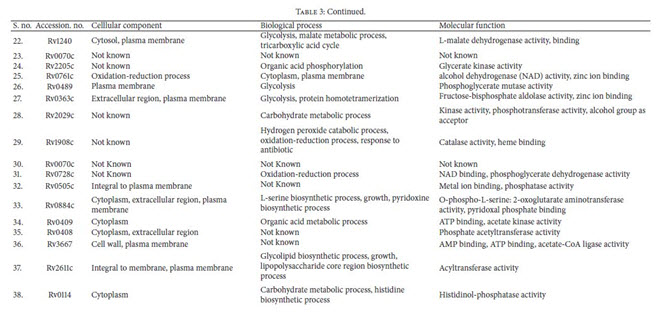
Identification of Essential Genes: Essential genes are thoseindispensable for the survival of an organism, and theirfunctions are, therefore, considered a foundation of life. Total55 enzymes out of all were found to be essential for M.tuberculosis life cycle (Table 2). These targets were found tobe potential targets and could be considered for rationaldrug design. Using metabolic pathway information as thestarting point for the identification of potential targets hasits advantages as each step in the pathway is validated as theessential function for the survival of the bacterium.
|
S. no. |
Entry no. |
Protein name |
Essential enzyme |
|
1. |
Rv1820 |
Acetolactate synthase |
Yes |
|
2 |
Rv0951 |
Succinyl-CoA synthetase subunit beta |
Yes |
|
3. |
Rv2987c |
Isopropylmalate isomerase small subunit |
Yes |
|
4. |
Rv1475c |
Aconitate hydratase (EC: 4.2.1.3) |
Yes |
|
5. |
Rv0066c |
Isocitrate dehydrogenase (EC: 1.1.1.42) |
Yes |
|
6. |
Rv2454c |
2-Oxoglutarate ferredoxin oxidoreductase subunit beta (EC: 1.2.7.3) |
Yes |
|
7. |
Rv1240 |
Malate dehydrogenase (EC: 1.1.1.37) |
Yes |
|
8. |
Rv1098c |
Fumarate hydratase (EC: 4.2.1.2) |
Yes |
|
9. |
Rv0247c |
Fumarate reductase iron-sulfur subunit (EC: 1.3.99.1) |
Yes |
|
10. |
Rv3356c |
Bifunctional 5,10-methylene-tetrahydrofolate dehydrogenase/5,10-methylene-tetrahydrofolate Cyclohydrolase (EC: 1.5.1.5 3.5.4.9) |
Yes |
|
11. |
Rv0951 |
Succinyl-CoA synthetase subunit beta (EC: 6.2.1.5) |
Yes |
|
12. |
Rv0904c |
Putative acetyl-coenzyme A carboxylase carboxyl transferase subunit beta (EC: 6.4.1.2) |
Yes |
|
13. |
Rv0973c |
Acetyl-/propionyl-coenzyme A carboxylase subunit alpha (EC: 6.3.4.14) |
Yes |
|
14. |
Rv1492 |
Methylmalonyl-CoA mutase small subunit (EC: 5.4.99.2) |
Yes |
|
15. |
Rv3667 |
Acetyl-CoA synthetase (EC: 6.2.1.1) |
Yes |
|
16. |
Rv0409 |
Acetate kinase (EC: 2.7.2.1) |
Yes |
|
17. |
Rv0408 |
Phosphate acetyltransferase (EC: 2.3.1.8) |
Yes |
|
18. |
Rv0243 |
Acetyl-CoA acetyltransferase (EC: 2.3.1.9) |
Yes |
|
19. |
Rv0860 |
Fatty oxidation protein FadB |
Yes |
|
20. |
Rv3667 |
Acetyl-CoA synthetase (EC: 6.2.1.1) |
Yes |
|
21. |
Rv0373c |
Carbon monoxyde dehydrogenase large subunit (EC: 1.2.99.2) |
No |
|
22. |
Rv2900c |
Formate dehydrogenase H (EC: 1.2.1.2) |
No |
|
23. |
Rv1023 |
Phosphopyruvate hydratase (EC: 4.2.1.11) |
Yes |
|
24. |
Rv1240 |
Malate dehydrogenase (EC: 1.1.1.37) |
Yes |
|
25. |
Rv0070c |
Serine hydroxymethyltransferase (EC: 2.1.2.1) |
Yes |
|
26. |
Rv2205c |
Hypothetical protein |
Yes |
|
27. |
Rv0761c |
Zinc-containing alcohol dehydrogenase NAD dependent AdhB (EC: 1.1.1.1) |
Yes |
|
28. |
Rv0489 |
Phosphoglyceromutase (EC: 5.4.2.1) |
Yes |
|
29. |
Rv0363c |
Fructose-bisphosphate aldolase (EC: 4.1.2.13) |
Yes |
|
30. |
Rv2029c |
Phosphofructokinase PfkB (phosphohexokinase) (EC: 2.7.1.—) |
Yes |
|
31. |
Rv1908c |
Catalase-peroxidase-peroxynitritase T KatG (EC: 1.11.1.6) |
Yes |
|
32. |
Rv0070c |
Serine hydroxymethyltransferase (EC: 2.1.2.1) |
Yes |
|
33. |
Rv0728c |
D-3-phosphoglycerate dehydrogenase (EC: 1.1.1.95) |
Yes |
|
34. |
Rv0505c |
Phosphoserine phosphatase (EC: 3.1.3.3) |
Yes |
|
35. |
Rv0884c |
Phosphoserine aminotransferase (EC: 2.6.1.52) |
Yes |
|
36. |
Rv0409 |
Acetate kinase (EC: 2.7.2.1) |
Yes |
|
37. |
Rv0408 |
Phosphate acetyltransferase (EC: 2.3.1.8) |
Yes |
|
38. |
Rv3667 |
Acetyl-CoA synthetase (EC: 6.2.1.1) |
Yes |
|
39. |
Rv2611c |
Lipid A biosynthesis lauroyl acyltransferase (EC: 2.3.1. —) |
Yes |
|
40. |
Rv0114 |
D-alpha,beta-D-heptose-1,7-biphosphate phosphatase (EC: 2. —.—.—) |
Yes |
|
41. |
Rv0113 |
Phosphoheptose isomerase (EC: 5.—.—.—) |
Yes |
|
42. |
Rv1315 |
UDP-N-acetylglucosamine 1-carboxyvinyltransferase (EC: 2.5.1.7) |
Yes |
|
43. |
Rv0482 |
UDP-N-acetylenolpyruvoylglucosamine reductase (EC: 1.1.1.158) |
Yes |
|
44. |
Rv2152c |
UDP-N-acetylmuramate-L-alanine ligase (EC: 6.3.2.8) |
Yes |
|
45. |
Rv2155c |
UDP-N-acetylmuramoyl-L-alanyl-D-glutamate synthetase (EC: 6.3.2.9) |
Yes |
|
46. |
Rv2157c |
UDP-N-acetylmuramoylalanyl-D-glutamyl-2,6-diaminopimelate-D-alanyl-D-alanyl ligase MurF |
Yes |
|
47. |
Rv2156c |
Phospho-N-acetylmuramoyl-pentapeptide-transferase (EC: 2.7.8.13) |
Yes |
|
48. |
Rv2153c |
Undecaprenyldiphospho-muramoylpentapeptide beta-N-acetylglucosaminyltransferase (EC: 2.4.1.227) |
Yes |
|
49. |
Rv2911 |
D-alanyl-D-alanine carboxypeptidase (EC: 3.4.16.4) |
No |
|
50. |
Rv2981c |
D-alanyl-alanine synthetase A (EC: 6.3.2.4) |
Yes |
|
51. |
Rv2136c |
Undecaprenyl pyrophosphate phosphatase (EC: 3.6.1.27) |
Yes |
|
52. |
Rv2911 |
D-alanyl-D-alanine carboxypeptidase (EC: 3.4.16.4) |
No |
|
53. |
Rv2158c |
UDP-N-acetylmuramoylalanyl-D-glutamate-2,6-diaminopimelate ligase (EC: 6.3.2.13) |
Yes |
|
54. |
Rv2157c |
UDP-N-acetylmuramoylalanyl-D-glutamyl-2,6-diaminopimelate-D-alanyl-D-alanyl ligase MurF |
Yes |
|
55. |
Rv2156c |
Phospho-N-acetylmuramoyl-pentapeptide-transferase (EC: 2.7.8.13) |
Yes |
|
56. |
Rv2153c |
Undecaprenyldiphospho-muramoylpentapeptide beta-N-acetylglucosaminyltransferase (EC: 2.4.1.227) |
Yes |
|
57. |
Rv3910 |
Transmembrane protein |
Yes |
|
58. |
Rv0016c |
Penicillin-binding protein PbpA |
Yes |
|
59. |
Rv2163c |
Penicillin-binding membrane protein PbpB |
Yes |
|
60. |
Rv2911 |
D-alanyl-D-alanine carboxypeptidase (EC: 3.4.16.4) |
Yes |
Table 2: Essential enzymes using DEG.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
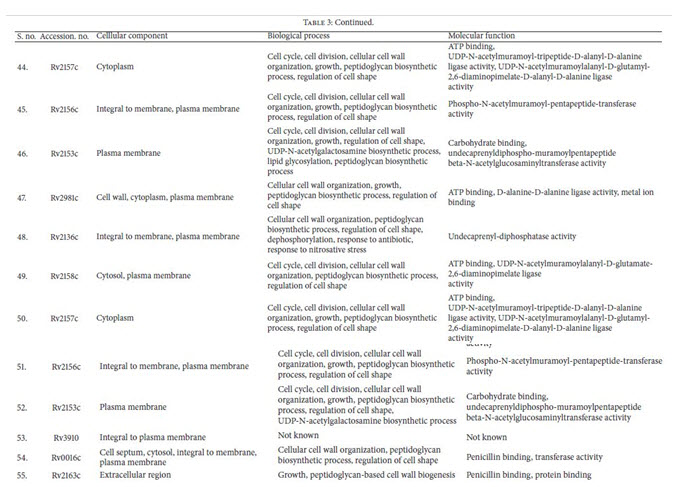
Identification of Drug Target’s Functions Using UniProt: The subcellular localization analysis of all supposed essentialand unique enzymes of M. tuberculosis were evaluatedby UniProt server. As it was suggested that, membraneassociated protein could be the better target for developingvaccines. After functional analysis unique enzymes involvedin cellular components like cell wall, cytoplasm, extracellularregion, plasma membrane, and so forth, their biologicalprocesses and their functions have been retrieved (Table 3).

CONCLUSION:
The metabolic pathways in the genome of Mycobacterium tuberculosisstrain Paris were analyzed and different putative targets were identified as unique drug targets.In the list of DEG result enzymes areinvolved in Peptidoglycan biosynthesis, Phosphotransferase system(PTS), DNA replication, ribosome, mismatch repair, protein exportand other many pathways. These identified putative targets may beexploiting further for developing drugs againstMycobacterium tuberculosis. the computational genomic approach hasfacilitated the search for potential drug targets against M.tuberculosis. Use of the DEG database is more efficient thanconventional methods for identification of essential genesand it facilitates the exploratory identification of the mostrelevant drug targets in the pathogen. It is quiteobvious that increase of drug resistance properties requires morepotential targets and by this Insilco approaches reduces the effort ofwet lab and also increases the probability of success. By this presentstudy we have tried to evaluate the targets could be better target forerational drug designing.
ACKNOWLEDGEMENT:
I would like to acknowledge Mr. K. Dhanabal for his inspiration and knowledge which made this work possible. I would also mention my parents and friends who have been constant support during the process of this work.
REFERENCE:
1. Sharma SK, Mohan V. Multidrug-resistant tuberculosis.Indian J Med Res 2004; 120 : 354-76.
2. Chakhaiyar P, Hasnain SE. Defining the mandate oftuberculosis research in a post genomic era. Med Princ Prac2004; 13 : 177-84.
3. S. H. E. Kaufmann, “Envisioning future strategies for vaccination against tuberculosis,” Nature Reviews Immunology, vol. 6, no. 9, pp. 699–704, 2006.
4. B. Greenwood, “Aglobal actionplan for the preventionandcontrol of pneumonia,” Bulletin of the World Health Organization, vol. 86, no. 5, p. 322, 2008.
5. P. Nunn, B. Williams, K. Floyd, C. Dye, G. Elzinga, and M. Raviglione, “Tuberculosis control in the era of HIV,” NatureReviews Immunology, vol. 5, no. 10, pp. 819–826, 2005.
6. Siddiqi N, Shamim M, Hussain S, Choudhary RK, Ahmed N, Prachee, et al. Molecular characterization of multidrugresistant isolates of Mycobacterium tuberculosis from patients in north India. Antimicrob Agents Chemother 2002;46 : 443-50.
7.Siddiqi N, Das R, Pathak N, Banerjee S, Ahmed N, Katoch VM, et al. Mycobacterium tuberculosis isolate witha distinct genomic identity overexpresses a TAP like efflux pump. Infection 2004; 32 : 109-11.
8. Majid AA, Ahmed N, Rao KR, Ghousunnissa S, Kausar F, Bose B, et al. AmpliBASE MT: A Mycobacteriumtuberculosis diversity knowledgebase. Bioinformatics 2002;20: 989-92.
9.A.M. Feist andB. Palsson, “Thegrowing scopeof applicationsof genome-scale metabolic reconstructions using Escherichia coli,” Nature Biotechnology, vol. 26, no. 6, pp. 659–667, 2008.
10. A. Dutta, S. K. Singh, P. Ghosh, R. Mukherjee, S. Mitter, and D. Bandyopadhyay, “In silico identification of potential therapeutic targets in the human pathogen Helicobacter pylori,” InSilico Biology, vol. 6, no. 1-2, pp. 43–47, 2006.
11. O. Ates, E. T. Oner, and K. Y. Arga, “Genome-scale reconstruction of metabolic network for a halophilic extremophile, Chromohalobacter salexigens DSM3043,” BMC Systems Biology, vol. 5, article 12, 2011.
12.A. Raghunathan, J. Reed, S. Shin, B. Palsson, and S. Daefler, “Constraint-based analysis of metabolic capacity of Salmonellatyphimurium during host-pathogen interaction,” BMC SystemsBiology, vol. 3, article 38, 2009.
13.A. M. Feist, C. S. Henry, J. L. Reed et al., “A genome-scale metabolic reconstruction for Escherichia coli K-12MG1655 that accounts for 1260 ORFs and thermodynamic information,” Molecular Systems Biology, vol. 3, article 121, 2007.
14. N. C. Duarte, M. J. Herrg˚ard, and B. Ø. Palsson, “Reconstruction and validation of Saccharomyces cerevisiae iND750, a fully compartmentalized genome-scale metabolic model,” GenomeResearch, vol. 14, no. 7, pp. 1298–1309, 2004.
15.A. K. Chavali, J.D.Whittemore, J. A. Eddy, K. T.Williams, and J. A. Papin, “Systems analysis of metabolism in the pathogenic trypanosomatid Leishmania major,” Molecular Systems Biology, vol. 4, article 177, 2008.
16. K. R. Sakharkar, M. K. Sakharkar, and V. T. K. Chow, “A novel genomics approach for the identification of drug targets in pathogens, with special reference to Pseudomonas aeruginosa,”
In Silico Biology, vol. 4, no. 3, pp. 355–360, 2004.
17. M. Y. Galperin and E. V. Koonin, “Searching for drug targets in microbial genomes,” Current Opinion in Biotechnology, vol. 10, no. 6, pp. 571–578, 1999.
18.M. Kanehisa, S. Goto, S. Kawashima, and A. Nakaya, “Thed KEGG databases at GenomeNet,” Nucleic Acids Research, vol. 30, no. 1, pp. 42–46, 2002.
19. R. Zhang, H.-Y. Ou, and C.-T. Zhang, “DEG: a database of essential genes,” Nucleic Acids Research, vol. 32, pp. D271–D272, 2004.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






