

 About Author: Shekh Yunus1, Nirmal Das Adhikary2 and Partha Chattopadhyay*
About Author: Shekh Yunus1, Nirmal Das Adhikary2 and Partha Chattopadhyay*
National Institute of Pharmaceutical Education and Research
at IICB, Jadavpur, 4 Raja S.C. Mullick Road, Kolkata-700032, India
Source of Support: NIPER Kolkata and IICB
ABSTRACT:
Click chemistry is a modular approach that uses only the most practical and reliable chemical transformations. Its applications are increasingly found in all aspects of drug discovery, ranging from lead finding through combinatorial chemistry and target-templated in situ chemistry, to proteomics and DNA research, using bioconjugation reactions. The copper-(I)-catalyzed 1,2,3-triazole formation from azides and terminal acetylenes is a particularly powerful linking reaction, due to its high degree of dependability, complete specificity, and the bio-compatibility of the reactants. The triazole products are more than just passive linkers; they readily associate with biological targets, through hydrogen bonding and dipole interactions.
[adsense:336x280:8701650588]
INTRODUCTION:
Click chemistry is a chemical philosophy introduced by K. Barry Sharpless of The Scripps Research Institute, in 2001 who got “NOBEL” prize in the same year. As defined by K. B. Sharpless “Click chemistry is a set of powerful, virtually 100% reliable, selective reactions for the rapid synthesis of new compounds via heteroatom links (C-X-C)…” It describes chemistry tailored to generate substances quickly and reliably by joining small units together. This is inspired by the fact that nature also generates substances by joining small modular units. Click chemistry is not a specific reaction; it is a concept that mimicks nature.
The laborious process of lead discovery and optimization has, in recent years, been aided by combinatorial chemistry, to generate collections of test compounds for screening. However, due to the large number of com-pounds that are involved, combinatorial chemistry is even more dependent than ‘traditional’ synthetic chemistry on the reliability of the individual reactions used to construct the new network of chemical bonds. Click chemistry is a new approach to synthesis that greatly facilitates this process making use of a few near-perfect chemical reactions for the synthesis and assembly of specially de-signed building blocks These building blocks have a high built-in energy content that drives a spontaneous and irreversible linkage reaction with complementary sites in other blocks.
The Click Chemistry Approach:
Click chemistry serves as a guiding principle in the quest for function: the search must be restricted to molecules that are easy to make. Focusing on lead discovery, this strategy
provides a means for the rapid exploration of the chemical universe. For lead optimization, it enables rapid SAR profiling, through generation of analog libraries. Click chemistry does not replace existing methods for drug discovery, but rather, it complements and extends them. It works well in conjunction with structure-based design and combinatorial chemistry techniques, and, through the choice of appropriate building blocks, can provide derivatives or mimics of ‘traditional’ pharmacophores, drugs and natural products. However, the real power of click chemistry lies in its ability to generate novel structures that might not necessarily resemble known pharmacophores.
Click chemistry is both enabled and con-strained by its reliance on a few nearly perfect reactions, and this, naturally, raises concerns about limitations on its access to chemical diversity. A computational study by Guida et al. suggests that the pool of ‘drug-like’ compounds (<30 non-hydrogen atoms, <500 Daltons; only H, C, N, O, P, S, F, Cl and Br; likely to be stable in the presence of water and oxygen) is as large as 1063. Currently, only a few million compounds (≈ 106–7) that fulfill these criteria are known, implying that only an infinitesimal part of the potential medicinal chemistry universe has been explored, to date. This has staggering implications for drug discovery. First and foremost, the vast majority of molecules with useful properties remain to be discovered. Second, most useful new compounds are likely to be found in unconventional structure space.
Click chemistry-based searches are fast, be-cause they avoid the regions of the ‘1063-uni-verse’ that are difficult to access. They are wide-ranging, owing to the use of strongly driven, highly selective reactions of broad scope, allowing a much greater diversity of block structures to be used. Thus, click chemistry makes the interesting proposition that greater diversity can be achieved with fewer reactions, because it is not the number of reactions that is important, but the tolerance of those reactions to variations in the nature of their components.
Defining a Click Chemistry Reaction:
Despite many successes, drug discovery approaches that are based on Nature’s secondary metabolites, ‘natural products,’ are often hampered by slow and complex syntheses Through the use of only the most facile and selective chemical transformations, click chemistry simplifies compound synthesis, providing the means for faster lead discovery and optimization. A click reaction must be of wide scope, giving consistently high yields with a variety of starting materials. It must be easy to perform, be insensitive to oxygen or water, and use only readily available reagents. Reaction work-up and product isolation must be simple, without requiring chromatographic purification.
How does one identify and then use such near-perfect reactions to accelerate the discovery of better and cheaper drug substances? Nature’s way in which she performs combinatorial chemistry serves as a source of inspiration: primary metabolism is highly modular – all proteins arise from 20 building blocks that are joined via reversible, heteroatom links (amides). Similarly, click chemistry uses carbon–heteroatom bond-forming connection chemistry. However, because we lack nature’s ability to perfectly control reversible carbonyl chemistry, we focus exclusively on highly energetic, ‘spring-loaded’ reactants and pure kinetic-control of the outcome. Thus, reversible carbonyl chemistry and irreversible click chemistry lie near opposite extremes of the continuum that connects pure thermodynamic and pure kinetic control
A focus on making carbon–heteroatom bonds must be accompanied by the use of pre-formed carbon–carbon bonds. The best, and most energetic, of these building blocks are olefins and acetylenes. Chemists have access to a plethora of such materials, ranging from naturally occur-ring terpenes to olefins from the petrochemical industry.
The Click Chemistry Universe:
A concerted research effort in laboratories and industries has yielded a set of extremely reliable processes for the synthesis of building blocks and compound libraries:
• Cycloaddition reactions, especially from the 1,3-dipolar family but also hetero-Diels-Alder reactions.
• Nucleophilic ring-opening reactions, especially of strained heterocyclic electrophiles, such as epoxides, aziridines, cyclic sulfates, cyclic sulfamidates, aziridinium ions and episulfonium ions.
• Carbonyl chemistry of the non-aldol type (e.g. the for-mation of oxime ethers, hydrazones and aromatic hete-rocycles).
• Addition to carbon–carbon multiple bonds; particularly oxidation reactions, such as epoxidation, dihydrox-ylation , aziridination , and nitrosyl and sulfenyl halide additions [13], but also certain Michael addition reactions.
It observed that the very best click reaction classes proceed most rapidly and in highest yield, not in water or water–co-solvent mixtures , but floating on water. For example, the 1,3-dipolar cycloadditions between diethyl acetylene dicarboxylate and diazido-cyclohexanediols proceed best in pure water .When the water is omitted so that the above reactants are mixed neat, the reactions are much slower and less selective, and, on a larger scale, become dangerous, be-cause click chemistry reactions are, by definition, highly exothermic. The presence of water in these reactions is beneficial, not just for reactivity reasons, but also because water is the best heat-sink for handling the enormous heat output when click reactions are performed on larger scales. Yet another advantage of water as a reaction solvent is that its presence prevents interference from simple protic func-tional groups, like alcohols and amides, which are ubiquitous in biologically active organic molecules.
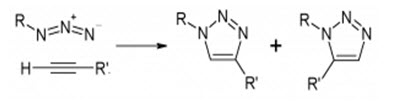
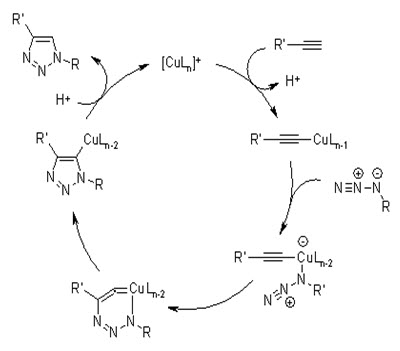
Huisgen’s 1,3-dipolar cycloaddition of alkynes and azides yielding triazoles is, undoubtedly, the premier example of a click reaction . Azides and alkynes are easy to install, and, despite being among the most energetic species known, they are also among the least reactive functional groups in organic chemistry. This stability, being purely kinetic in origin, is responsible for the slow nature of the cycloaddition reaction and the inertness of these functional groups towards biological molecules and towards the reaction conditions inside living systems (i.e. aqueous and mild reducing environments). With the recently discovered dramatic rate acceleration of the azide–alkyne coupling event under copper-(I) catalysis, and the beneficial effects of water, this unique connection process seems to be redefining the concept of ‘a perfect’ reaction. Given this new process, and the ready availability of the starting materials, highly diverse, unambiguous libraries become available quickly. There are no protecting groups, and with complete conversion and selectivity for the 1,4-disubstituted 1,2,3-triazole, structural uncertainties do not exist, rendering purification unnecessary.
The transformation is especially relevant for drug discovery, not just because of its reliability as a linking reaction, but also because of the favourable physicochemical proper-ties of triazoles. The latter serve as rigid linking units that place the carbon atoms, attached to the 1,4-positions of the 1,2,3-triazole ring, at a distance of 5.0 Å (C-α distance in amides: 3.8 Å). In contrast to amides, triazoles cannot be cleaved hydrolytically or otherwise, and unlike benzenoids and related aromatic heterocycles, they are almost impossible to oxidize or reduce. They possess a large dipole moment of ~5 Debye and nitrogen atoms two and three function as weak hydrogen bond acceptors.
Click Chemistry and Drug Discovery:
Click chemistry is being used increasingly in biomedical research, ranging from lead discovery and optimization, to tagging of biological systems, such as proteins, nucleotides and whole organisms. The potential of this approach is highlighted here, by reviewing several early applications.
Synthesis of Lead Discovery Libraries:
Over the course of five years, click chemistry laboratories at Coelacanth Corporation (now Lexicon Pharmaceuticals https://www.lexicon-genetics.com/pharma) have employed solution-phase chemistry to produce a variety of screening libraries, containing a total of 200 000 individual com-pounds, each more than 85% pure and available in 25–50 mg amounts. In line with the click chemistry philosophy, each library compound was produced in only one or two synthetic steps, from key building block reagents, using automated liquid handling workstations. Despite the short synthetic sequences, much compound diversity and novelty was achieved by starting with non-commercial building block reagents, prepared in-house on multi-gram or, even, kilogram scales. Examples include ‘spring-loaded’ epoxides and aziridines for the formation of 1,2-difunc-tionalized compounds by nucleophilic opening , imidoesters for the generation of five-membered aromatic heterocycles , azides for the synthesis of 1,2,3-triazole-derived libraries via 1,3-dipolar cycloaddition with β - ketoesters , and 3-aminoazetidines for the preparation of non-aromatic heterocyclic libraries. Also, targeted libraries were made, one of which led to the discovery of potent Peroxisome Proliferator-Activated Receptor γ (PPAR-γ ) agonists.
The copper-(I)-catalyzed formation of 1,2,3-triazoles has recently been used to prepare functionalized resins for the solid phase synthesis of a library of dopaminergic arylcarbamides. In another resin-based approach, Yli-Kauhaluoma et al. prepared 1,2,3-triazoles via thermal 1,3-dipolar cycloaddition of polymer-bound azides to alkynes, followed by cleavage from the solid support with TFA.
Drug discovery approaches based on copper-(I)-catalyzed formation of triazole from azides and acetylenes: Examples include (a) synthesis of neoglycoconjugates (b) synthesis of fucosyltransferase inhibitors (c) Developement of HIV protease inhibitors
In Situ Click Chemistry:
Mock’s discovery of a dramatic rate acceleration of the azide–alkyne cycloaddition by sequestering the two com-ponents inside a host structure, prompted Sharpless et al. to investigate a new paradigm for drug discovery, which is dependent on irreversible, target-guided synthesis of high-affinity inhibitors from reagents that are inert under physiological conditions. By contrast, other ap-proaches employ highly reactive reagents (e.g. aldehydes and hydrazines; thiols and α -chloroketones etc.) and reversible reactions for the in situ assembly of inhibitors inside a target’s binding pocket. Acetylcholine es-terase (AChE) was chosen as the target. Its inhibitors have been employed for over a century in various therapeutic regimens and to investigate the role of acetylcholine in neurotransmission. The enzyme’s active site is lo-cated at the base of a narrow gorge, ~20 Å in depth. A second, peripheral binding site exists at the rim of the gorge, near to the enzyme surface. The concerted thermal 1,3-dipolar cycloaddition reaction between azide and acetylene reagents (which carry active-site and periph-eral-site binding groups via flexible spacers) was selected for this study for several reasons. First, the reaction is extremely slow at room temperature, second, it does not involve components that might disturb the binding sites (external reagents, catalysts, by-products), and third, the reactants are bio-orthogonal. A substantial rate accelera-tion was observed for certain azide–acetylene reagent combinations in the presence of the enzyme. From 49 building block combinations, the enzyme selected the TZ2/PA6 pair, leading to the formation of a sole reaction product in a highly regioselective fashion: the TZ2PA6 syn-6 triazole. By contrast, chemical synthesis in the absence of enzyme provided a roughly 1:1 mixture of syn-and anti-6 regioisomers. Both are respectable inhibitors, but the syn-6 isomer, with a 100-fold greater affinity and a sub-picomolar dissociation constant for certain acetyl-choline esterases, has a potency greater than all known non-covalent organic AChE inhibitors. Thus, AChE itself, served as the reaction vessel, synthesizing its own inhibitor by equilibrium-controlled sampling of various possible pairs of reactants in its gorge until the irreversible cycload-dition between azide and acetylene essentially ‘froze’ the pair that fits best into the binding pocket. A recent X-ray crystallographic analysis of both the syn-6- and anti-6-mouse AChE complexes at 2.45–2.65 Å resolution, reveals that the former has effectively trapped the enzyme in a previously unknown conformational state. If this ‘open’ conformer is in facile equilibrium with the ground state, it might help to explain the enormous turnover rates of acetylcholine esterases – similar to a breathing motion that is in pace with the catalytic cycle. In addition, this study revealed that the 1,2,3-triazole cores interact strongly with the protein through hydrogen bonding to N2 and N3 and through their large dipole moments (~5 Debye).
Click Chemistry and Bioconjugation:
In vivo and in vitro bioconjugation applications benefit from the unprecedented reliability of the copper-catalyzed azide–acetylene union, the inertness of the reactants under physiological conditions, and the mild reaction conditions.
Tagging of Live Organisms and Proteins:
The application of mild methods for the chemical modifi-cation of components in, or on, living cells under physio-logical conditions has been pioneered by the Bertozzi group. Finn et al. recently succeeded in using the new copper-(I)-catalyzed 1,2,3-triazole formation for label-ing intact Cowpea mosaic virus particles (CPMV) with fluorescein. CPMV’s capsid is made up of 60 copies of an asymmetric two-protein unit, which encapsu-late the single-stranded RNA genome. Each unit contains one reactive Cys and Lys. To these, via traditional bioconjugation methods, a total of 60 azides per virus particle were attached. Under optimized conditions, all 60 azide groups reacted to form triazoles, for a yield of the 60-mer of >95%.
Tirrell and Link have recently disclosed how, using an ingenious sequence of molecular biology techniques, they forced Eschericia coli cells into making and displaying an azide-bearing outer membrane protein C (OmpC), in which methionine residues were replaced by azidohomoalanine 7. These bacterial cells, now presenting azide groups to the extracellular milieu, were successfully biotinylated, under the special copper-catalyzed conditions developed by the Finn group for their virus case, with a biotin-alkyne reagent.
The Schultz laboratory then reported that their method for genetically-encoded incorporation of the azide and acetylene tyrosine analogs 8 and 9 into proteins of Saccharomyces cerevisiae, could be followed up by capture of dyes, using the copper-(I)-catalyzed azide–alkyne tria-zole-coupling, as optimized by Finn et al. for in situ bioconjugations.
Activity-Based Protein Profiling (ABPP):
ABPP is a chemical method that employs active site-directed probes to tag proteins and monitor their expression levels and function in complex proteomes; however, the bulky reporter tags that are currently used require cell homogenization before analysis, thereby, preventing measurements in living organisms. Cravatt et al. have solved this problem with small, cell-permeable reagents that carry an azide group for later dye attachment via the bio-orthogonal, copper-(I) catalyzed reaction with acetylenes. The authors detected glutathione S-transferases (GSTO 1–1), aldehyde dehydrogenases (ALDH-1) and enoyl CoA hydratases (ECH-1) at endogenously expressed levels in viable cells, by incubation with 6-azidohexyl benzene-sulfonate (PS-N3), followed by homogenization and tagging with Rhodamine-acetylene (Rh-acetylene). Quantification of GSTO 1–1 levels in breast cancer cell lines yielded results that were comparable with traditional methods. The Cravatt approach works even in live animals, allowing ECH-1 to be observed in the heart muscle of mice one hour after injection with PS-N3 and ‘staining’ of the unique protein from the crude heart-homogenate, via copper-catalyzed conjugation with Rh-acetylene. By enabling the determination of protein expression levels in living organ-isms, this new in vivo ABPP method provides more unbiased results, because tagging occurs before cell death.
Labelling of DNA:
Ju et al. prepared primers for the Sanger dideoxy chain ter-mination reaction for DNA sequencing, by tagging the M13–40 universal forward sequencing primer at its 5′-end, with alkynyl 6-carboxyfluorescein (FAM), using the con-certed thermal 1,3-dipolar cycloaddition process (Figure 4c) [60]. The advantages of this method over existing ones are that it proceeds in high yield under relatively mild conditions, without the need for additives (e.g. cata-lysts or reagents) or the formation of by-products, rendering purification of the labeled single-stranded DNA unneces-sary. The FAM-triazole-M13–40 primer was successfully used in the Sanger method, terminating with biotinylated dideoxyATP (ddATP-Biotin), to produce DNA sequencing fragments, using PCR-amplified DNA as a template. The DNA fragments were analyzed by capillary array elec-trophoresis, the peaks representing the FAM fluorescence emission from each DNA fragment that was extended from the fluorescence-labeled primer and terminated by ddATP. The ‘A’ sequencing ladder that was obtained in this way matched, exactly, the sequence of the DNA template.

Fig: click chemistry and Bioconjugation
Conclusions:
In summary, click chemistry has proven to be a powerful tool in biomedical research, ranging from combinatorial chemistry and target-templated in situ chemistry for lead discovery, to bioconjugation strategies for proteomics and DNA research. Of the various click chemistry reactions that are available to us, the union of azides and acetylenes to give triazoles deserves special recognition. Azides and acetylenes are stable across a broad range of organic reac-tion conditions and in biological environments, yet they are highly energetic functional groups. Their irreversible combination to triazoles is highly exothermic, albeit slow. The full potential of this ligation reaction was unleashed with the discovery of copper-(I) catalysis. Benefiting from more than a million-fold rate acceleration, this process proceeds in near-quantitative yields in water, and because no protecting groups are used, the products are screened directly from the reaction mixture. This triazole forming process, and click chemistry in general, promise to acceler-ate both lead finding and lead optimization, due, above all, to its great scope, modular design, and reliance on extremely short sequences of near-perfect reactions.
References:
1) Kolb, H.C. et al. (2001) Click chemistry: diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. Engl. 40, 2004–2021
2) Sneader, W. (1996) Drug Prototypes and Their Exploitation, John Wiley & sons
3) Bemis, G.W. and Murcko, M.A. (1996) The properties of known drug 1. Molecular frameworks. J. Med. Chem. 39, 2887–2893
4) Bohacek, R.S. et al. (1996) The art and practice of structure-based drug design: a molecular modeling perspective. Med. Res. Rev. 16, 3–50
5) Newman, D.J. et al. (2003) Natural products as sources of new drugs over the period 1981-2002. J. Nat. Prod. 66, 1022–1037
6) Shu, Y-Z. (1998) Recent natural products based drug development: a pharmaceutical industry perspective. J. Nat. Prod. 61, 1053–1071
7) Huisgen, R. (1984) 1,3-Dipolar cycloaddition – introduction, survey, mechanism. In 1,3-Dipolar Cycloaddition Chemistry (Vol. 1) (Padwa, A., ed.), pp. 1-176, Wiley
8) Jorgensen, K.A. (2000) Catalytic asymmetric hetero-Diels-Alder reactions of carbonyl compounds and imines. Angew. Chem. Int. Ed. Engl. 39, 3558–3588
9) Tietze, L.F. and Kettschau, G. (1997) Hetero Diels-Alder reactions in organic chemistry. Top. Curr. Chem. 189, 1–120
10) Adolfsson, H. et al. (1999) Comparison of amine additives most effective in the new methyltrioxorhenium-catalyzed epoxidation process. Tetrahedron Lett. 40, 3991–3994
11) Kolb, H.C. et al. (1994) Catalytic asymmetric dihydroxylation. Chem. Rev. 94, 2483–2547
12) Gontcharov, A.V. et al. (1999) tert-Butylsulfonamide. A new nitrogen source for catalytic aminohydroxylation and aziridination of olefins. Org. Lett. 1, 783–786
13) Kühle, E. (1970) One-hundred years of sulfenic acid chemistry. IIa. Oxidation, reduction, and addition reactions of sulfenyl halides. Synthesis (Mass.) 11, 563–586
14) Gholami, M.R. and Yangjeh, A.H. (1999) Hydrophobic effects in 1,3- dipolar cycloaddition of C,N-diphenylnitrone with Dibutyl fumarate in aqueous solutions. J. Chem. Res. 3, 226–227
15) Lee, L.V. et al. (2003) A potent and highly selective inhibitor of human a-1,3-Fucosyltransferase via click chemistry. J. Am. Chem. Soc. 125, 9588–9589
16) Rostovtsev, V.V. et al. (2002) A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective ‘ligation’ of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 41, 2596–2599
17) Torne, C.W. et al. (2002) Peptidotriazoles on solid phase: [1,2,3]- Triazoles by Regiospecific Copper(I)-catalyzed 1,3-Dipolar Cycloadditions of terminal Alkynes to Azides. J. Org. Chem. 67, 3057–3064
18) Purcell, W.P. and Singer, J.A. (1967) Electronic and molecular structure of selected unsubstituted and dimethyl amides from measurements of electric moments and nuclear magnetic resonance. J. Phys. Chem. 71, 4316–4319
19) Kolb, H.C. (2001) Application of Click Chemistry to the generation of new chemical libraries for drug discovery. In 221st Am. Chem. Soc. Meeting, Abstr. Pap. ORGN-434
20) Kolb, H.C. et al. (2003) Large scale synthesis of optically pure aziridines US2003153771
21) Kolb, H.C. et al. (2003) Modified safe and efficient process for the environmentally friendly synthesis of imidoesters by Pinner addition using hydrogen chloride solutions. US2003153728
22) Kolb, H.C. et al. (2003) Preparation of 1,2,3-triazole carboxylic acids from azides and β -ketoesters in the presence of base. US2003135050
23) Chen, Z. et al. (1999) Preparation of 3-aminoazetidines as building blocks for combinatorial libraries. WO 9919297(A1)
24) Kolb, H.C. et al. (2000) Preparation of PPAR-(gamma) agonists as agents for the treatment of type II diabetes. WO2000063161
25) Löber, S. et al. (2003) Click linker: efficient and high-yielding synthesis of a new family of SPOS resins by 1,3-dipolar cycloaddition. Org. Lett. 5, 1753–1755
26) Harju, K. et al. (2003) Solid-phase synthesis of 1,2,3-triazoles via 1,3- dipolar cycloaddition. J. Comb. Chem, (in press)
27) (2003) Carbohydrate-based Drug Discovery. (Wong, C.-H., ed.), pp. 980, Wiley-VCH
28) Mammen, M. et al. (1998) Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. Engl. 37, 2755–2794
29) Perez-Balderas, F. et al. (2003) Multivalent neoglycoconjugates by regiospecific cycloaddition of alkynes and azides using organic-soluble copper catalysts. Org. Lett. 5, 1951–1954
30) Sears, P. and Wong, C.H. (1998) Enzyme action in glycoprotein synthesis. Cell. Mol. Life Sci. 54, 223–252
31) Compain, P. and Martin, O.R. (2001) Carbohydrate mimetics-based glycosyltransferase inhibitors. Bioorg. Med. Chem. 9, 3077–3092
32) Mitchell, M.L. et al. (2002) Synthesis and evaluation of transition-state analogue inhibitors of a-1,3-fucosyltransferase. Angew. Chem. Int. Ed. Engl. 41, 3041–3044
33) Abdel-Rahman, H.M. et al. (2002) HIV protease inhibitors: peptidomimetic drugs and future perspectives. Curr. Med. Chem. 9, 1905–1922
34) Veronica Miller (2001) Resistance to protease inhibitors. J. Acquir. Immune Defic. Syndr. 26(Suppl. 1), S34–S50
35) Brik, A. et al. A rapid diversity-oriented synthesis and in situ screening of HIV protease inhibitors. Chembiochem (in press)
36) Tung, R.D. et al. (2002) Design and synthesis of amprenavir, a novel HIV protease inhibitor. Infectious Disease and Therapy 25, 101–118
37) Mock, W.L. et al. (1983) Cycloaddition induced by cucurbituril. A case of Pauling principle catalysis. J. Org. Chem. 48, 3619–3620
38) Mock, W.L. et al. (1989) Catalysis by cucurbituril. The significance of bound-substrate destabilization for induced triazole formation. J. Org. Chem. 54, 5302–5308
39) Mock, W.L. (1995) Cucurbituril. Top. Curr. Chem. 175, 1–24
40) Lewis, W.G. et al. (2002) Click chemistry in situ: Acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array of building blocks. Angew. Chem. Int. Ed. Engl. 41, 1053–1057
41) Rideout, D. (1986) Self-assembling cytotoxins. Science 233, 561–563
42) Rideout, D. et al. (1990) Synergism through direct covalent bonding between agents: a strategy for rational design of chemotherapeutic combinations. Biopolymers 29, 247–262
43) Nguyen, R. and Huc, I. (2001) Using an enzyme’s active site to template inhibitors. Angew. Chem. Int. Ed. Engl. 40, 1774–1776
44) Lehn, J-M. and Eliseev, A.V. (2001) Dynamic combinatorial chemistry. Science 291, 2331–2332
45) Argyl-Robertson, D. (1863) The Calabar bean as a new agent in ophthalmic practice. Edinburgh Med. J. 8, 815–820
46) Dale, H.H. (1914) The action of certain esters of choline and their relation to muscarine. J. Pharmacol. Exp. Ther. 6, 147–190
47) Sussman, J.L. et al. (1991) Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science 253, 872–879
48) Changeux, J.P. (1966) Responses of acetylcholinesterase from Torpedo marmorata to salts and curarizing agents. Mol. Pharmacol. 2, 369–392
49) Taylor, P. and Lappi, S. (1975) Interaction of fluorescence probes with acetylcholinesterase. The site and specificity of propidium binding. Biochemistry 14, 1989–1997
50) Bourne, Y. et al. (2003) Structural insights into ligand interactions at the acetylcholinesterase peripheral anionic site. EMBO J. 22, 1–12
51) Harel, M. et al. (1993) Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. U. S. A. 90, 9031–9035
52) Mahal, L.K. et al. (1997) Engineering chemical reactivity on cell surfaces through oligosaccharide biosynthesis. Science 276, 1125–1128
53) Click chemistry: Diverse Chemical function from A Few Good Reactions,H.C. kolb,M.G.Finn and K.B. Sharpless, Angew.chem ,Int.ed. 2001,40,pp. 2001-2021
54) Click chemistry: The growing impact of click chemistry on drug discovery Hartmuth C. Kolb and K. Barry Sharpless review article ,DDT Vol. 8, No. 24, December 2003
55) Chinmay chowdhury,Anup kumar sasmal,P.K.Dutta, Tetrahedron Letters 2009 (50, 2678-2681)
56) K.C. Majumdar, Krishnau Ray, Sintu Ganai, Synthesis 2010,No.12,pp 2101-2105
57) K.C. Majumdar, Krishnau Ray, Sintu Ganai,Tapas Ghosh, Synthesis 2010,No.5,pp 0858-0862
Reference ID: PHARMATUTOR-ART-1035
FIND OUT MORE ARTICLES AT OUR DATABASE






