

 About Author:
About Author:
ABHISHAK SINGH PARMAR,
M.PHARMACY (MEDICINAL AND PHARMACEUTICAL CHEMISTRY),
B.R.NAHATA COLLEGE OF PHARMACY,
MANDSAUR (M.P).
*abhishakparmar@gmail.com
1. INTRODUCTION
1.1 Medicinal and Pharmaceutical chemistry
It is a discipline at the intersection of chemistry and pharmacology involved with designing, synthesizing and developing pharmaceutical drugs. Medicinal chemistry involves the identification, synthesis and development of new chemical entities suitable for therapeutic use. It also includes the study of existing drugs, their biological properties, and their quantitative structure-activity relationships (QSAR). Pharmaceutical chemistry is focused on quality aspects of medicines and aims to assure fitness for the purpose of medicinal products. Compounds used as medicines are overwhelmingly organic compounds including small organic molecules and biopolymers. However, inorganic compounds and metal-containing compounds have been found to be useful as drugs. For example, the cis-platin series of platinum-containing complexes have found use as anti-cancer agents.1
Medicinal chemistry is a highly interdisciplinary science combining organic chemistry with biochemistry, computational chemistry, pharmacology, pharmacognosy, molecular biology, statistics, and physical chemistry.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-1629
Discovery
The first step of drug discovery involves the identification of novel active compounds, often called "hits", which are typically found by screening many compounds (compound library) for the desired biological properties. While a number of approaches toward the identification of hits exist, the most successful of techniques relies on chemical and biological intuition developed through years of rigorous chemical-biological training. Other sources of hits can come from natural sources, such as plants, animals, or fungi. Hits may originate also from random chemical libraries, such as those created through combinatorial chemistry or historic chemical compound collections that are tested en-masse against the biological target in question.1
Optimization
Another step in drug discovery involves further chemical modifications in order to improve the biological and physiochemical properties of a given candidate compound library. Chemical modifications can improve the recognition and binding geometries (Pharmacophore) of the candidate compounds, their affinities and pharmacokinetics, or indeed their reactivity and stability during their metabolic degradation. The quantitative structure-activity relationship (QSAR) of the Pharmacophore play an important part in finding lead compounds, which exhibit the most potency, most selectivity, best pharmacokinetics and least toxicity. QSAR involves mainly physical chemistry and molecular docking tools (CoMFA and CoMSIA), that leads to tabulated data and first and second order equations. There are many theories, the most relevant being Hansch's analysis that involves Hammett electronic parameters, steric parameters and logP (lipophilicity) parameters.1
Development
The final step involves the rendering the lead compounds suitable for use in clinical trials. This involves the optimization of the synthetic route for bulk production, and the preparation of a suitable drug formulation.1
1.2 Drug design concept
Paul eldritch provided rationality to modern drug research. “Prontosil” an antibacterial agent provided the concept of drug design to medicinal chemist. The drug design is based mainly on the modification of lead molecule which suffers from many unwanted side effects. The efficiency of a lead molecule can be increased by High through out screening (HT.P.S) technique with utilizes cell line culture system with Enzyme linked immune system assay (E.L.I.S.A.) and receptors molecules which are derived from gene cloning.
The new techniques like molecular graphics and computational chemistry provides new chemical structure which helps in making potent new drugs.2
The various stages covered in medicinal chemistry are:-
(1) First stage involves the identification of new active compounds known as lead molecules. The lead molecules are derived from natural substances, organic chemical reactions and biotechnological process.
(2) In the second stage the lead molecule is further optimized to prepare a compound with improved biological activity, selectivity, potency and reduced toxicity.
(3) The third stage involves optimization of method of production to produce drug in bulk amount and further modification of pharmacokinetic and pharmaceutical properties of active substance to make it a clinically useful compound.
1.3 Hypertension
Hypertension is defined as condition in which systolic and or diastolic blood pressure exceed above 140/90mm Hg, an agent that lowers blood pressure is called the antihypertensive agent. The most desired action is slow reduction of blood pressure with prolonged effect further increased doses should cause a more prolonged effect rather than a more pronounced fall in blood pressure finally the drug should be active after oral administration because they would be used for extended periods.3
In today’s era of globalization, characterized by hurry, worry, and curry, the incidences of cardiovascular diseases are on the rise. Hypertension is a condition where the blood pressure is constantly higher than normal. This poses a serious health risk because it forces the heart to work extra hard. Constantly higher blood pressure can damage the coronary arteries, the brain, the kidneys, and the eyes. Hypertension is a major cause of strokes and heart attacks.4
· Types of Hypertension
(1)Primary hypertension
Despite many years of active research, there is no single major factor that can be attributed to primary hypertension. There is a natural progression of the disease which suggests that an early elevation in blood volume and cardiac output might initiate subsequent changes in systemic vasculature (increased resistance).
Though the specific cause for this type is unknown, almost 90% of the total hypertension cases are of this type. It is also called as ‘essential or idiopathic’ hypertension.4
(2) Secondary hypertension
There are many known conditions that can cause secondary hypertension also known as ‘inessential hypertension’. Regardless of the cause, arterial pressure becomes elevated either due to increase in cardiac output or increase in systemic vascular resistance or both. When cardiac output is elevated, it is generally due to either increased neurohumoral activation of the heart or increased blood volume. The cause may be any of these: renal artery disease, eclampsia of pregnancy or pheochromocytoma. Only around 10% of the total cases belong to this category.4
· Causes and pathological risks of uncontrolledHypertension
High blood pressure is a potential risk factor for cardiovascular diseases, independent of the presence or absence of other risk factors, for example, smoking,diabetes, and/or hypercholesterolemia. The other factorssuch as genetics, age, sex, race, diet, and environmentalfactors (e.g., stress and physical activity), also playimportant role in elevation of the blood pressure.
· Etiology of hypertension
However the hypertension may arise secondary to many diseases but the hypertension is known to be due to genetic history of the family. If the parents suffer from hypertension then in children hypertension may occur. It is seen that more than 90% of the black people are suffered from hypertension than whites. Thus it is found more in black peoples. It occurs more in middle age men than in middle age women. Environmental factors like working in stressful lifestyle, excess sodium intake, obesity, and smoking, all further increases chances of hypertension.5
· Control of blood pressure
Blood pressure in its simplest term is the force of pumping of heart action, working against the resistance provided by the blood vessels. Body has a special ‘blood pressure system’. The purpose of this system is to maintain blood flow to all the tissues of the body at rest or during movements (figure-1). Table -1 depict the delicate coordination between cardiovascular system and the sympathetic nervous system involving some important organs like kidneys, brain, adrenals, and heart, to regulate the blood pressure. Genetic and environmental factors cause imbalance in this and cause hypertension.4
· Mechanism for controlling blood pressure
Arterial blood pressure is controlled with a narrow range to provide a sufficient perfusion of the tissues without causing damage to the vascular system, specially the arterial intimae. The arterial blood pressure is directly proportional to the product of cardiac output and peripheral vascular resistance. In normal and abnormal hypertensive individual both the cardiac output and peripheral resistance are regulated mainly by two overlapping controlled mechanisms. In controlling of blood pressure Benzothiazepine and thiosalicylamide analogous play important role by the blocking calcium channel present in blood vessels. The baroreflexes are mediated by the sympathetic nervous system and the rennin angiotensin- aldosteron system. Most of the antihypertensive drugs lower the blood pressure by reducing cardiac output, or decreasing peripheral resistance.
1.4 Antihypertensive drugs
History and classification.
The discovery of most of our current antihypertensive drugs did not involve the target design of molecules to modify a blood pressure control system. Most of the drugs have evolved out through conventional synthesis and biological evaluation processes based on QSAR studies.The currently used antihypertensive drugs are classified into seven broad categories.4
Table no.1 Classes of currently established drugs for the treatment of hypertension
|
Class |
Chemical |
Examples of drugs |
|
Diuretics |
Thiazide type
Potassium sparing type Looptype |
Chlorothiazide, Chlorthalidone, Bendroflumethiazide, Trichlormethiazide Spironolactone, Amiloride, Triamterene Furosemide, Ethacrynic acid, Bumetanide, Torasemide |
|
Β-Blockers |
Non-selective (β1/ β2)
Selective β1 |
Propranolol, Timolol, Nadolol, Pindolol, Cartetolol Atenolol, Betaxolol, Metaprolol, Acebutolol, Tozalol |
|
ACE inhibitors |
|
Captopril, Enalapril, Lisinopril, Fosinopril, Ramipril |
|
Ca2+ blockers |
Dihydropyridine, Phenyalkylamines Verapamil, Gallopumil, Benzothiazepines, thosalicylamide. |
Nifedipine, Nicardipine, Felodipine, Amlodipine Diltiazem, Thisalicylamide derivative |
|
α1-Adr antagonists |
|
Prazosin, Doxazosin, Terazosin, Labetolol |
|
α2-Adr agonists |
|
Clonidine, Guanafexine, Guanabenz |
|
Miscellane-ous |
|
α-Methyldopa,Euronalblockers(Bretilium, Guanethedine),Rauwolfia and its alkaloids, (Reserpine, Deserpine, Rauwolfia whole root),Ganglionic blockers (Guanadrel, Mecamylamine, Hexamethonium), Non-specific vasodilators (Hydralazine, Nitroprusside, Diazoxide, Minoxidil. |
1.5 Calcium channels
Voltage-gated calcium channels are integral membrane proteins that allow calcium ions to flow into the cell cytoplasm from the extracellular milieu, in response to membrane depolarization. This class of ion channel is found in virtually all types of excitable cells, ranging from neurons and glial cells to muscle cells. The functional inventory of calcium channels is equally broad, spanning from triggering of muscle contraction over control of neurotransmitter release to electrical excitation (calcium action potentials). The diversity of calcium channel function is reflected in their molecular heterogeneity: Several genes, encoding biophysically and pharmacologically distinct types of calcium channels, have been cloned and characterized functionally. These different types of calcium channels play specialized roles in cellular function; for instance, L-type calcium channels mediate muscle contraction and N- and P/Q-type calcium channels control neurotransmitter release.12
[adsense:468x15:2204050025]
· Regulation of intracellular calcium levels
Most of the calcium ions in a resting cell is sequestered in organelles, particularly the endoplasmic or sarcoplasmic reticulum (ER or SR) and the mitochondria , and the free calcium ion is kept to a low level, about 10-7 M. The calcium ionsconcentration in tissue fluid is about 2.4 Mm, so there is a large concentration gradient favouring Ca2+ entry. Free calcium ion is kept low (a) by the operation of active transport mechanisms that eject cytosolic Ca2+ through the plasma membrane and pump it into the ER , and (b) by the normally low Ca2+ permeability of the plasma and ER membrane. Regulation of free calcium ion involves three main mechanisms: 1. Control of Ca2+ entry
2. Control of Ca2+ extrusion
3. Exchange of Ca2+ between the cytosol and the intracellular stores.7
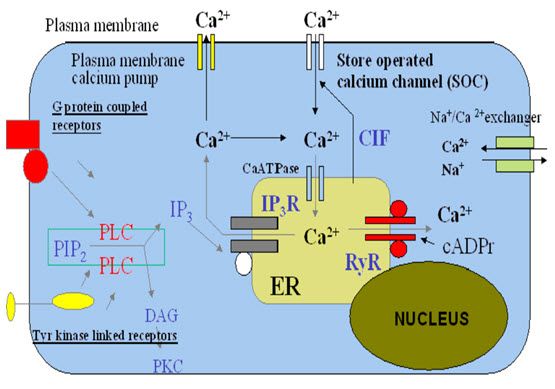
Figure -2 Regulation of intracellular calcium7
Calcium entry mechanism There are four main routes by which Ca+2 enter cells across the plasma membrane:
I.Voltage-gated calcium channel
II. Ligand-gated calcium channel
III. Store–operated calcium channel
IV. Na+- Ca+2 exchange.7
I. Voltage- gated calcium channel
Voltage-gated calcium channels are integral membrane proteins that allow calcium ions to flow into the cell cytoplasm from the extracellular environment, in response to membrane depolarization.In excitable cells, voltage-dependent calcium channels (VDCCs) are responsible for the increase of intracellular free calcium concentration, by the influx of extracellular Ca+2 across the plasma membrane .In non-excitable cells, an increase of intracellular free calcium concentration results: (i) from the activation of intracellular Ca+2 stores, such as endoplasmatic reticulum, and (ii) from a receptor-mediated Ca+2 entry across the plasma membrane.8
Voltage-gated calcium channel have been classified their electrophysiological and pharmacological properties and are generally divided into low-threshold (T-type) and high threshold (L-, N-, P/Q- and R-types). The L-, N-, P/Q- and R-type channels typically activate at membrane potentials near - 30 mV and display diverse kinetic, voltage-dependent and pharmacological properties.12
The subtypes vary with respect to their activation and inactivation kinetics, their conductance, and their voltage threshold for activation, their conductance and their sensitivity to blocking agents as summarized in table1(rang and dales).These different types of calcium channels play specialized roles in cellular function; for instance, L-type calcium channels mediate muscle contraction and N- and P/Q-type calcium channels involved in neurotransmitter release and T type channel mediate Ca+2 dependent functions such as regulation of other channel ,enzyme etc.7
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
T-type calcium channel
The T-type calcium channel is a type of voltage-gated calcium channel. "T" stands for transient referring to the length of activation. T-type calcium channel mediate Ca+2 entry into neurons and thereby control various Ca+2 dependent function such as regulation of other channel, enzyme, etc. . The other sub-types of voltage-gated calcium channel, the α1 subunit is the one that determines most of the channel's properties
T-type calcium channels may contain one of three α1 subunits, α1G (Cav3.1), α1H (Cav3.2) or α1I (Cav3.3).
Along with sodium "funny current," the T-type calcium channel produces the pacemaker potential in the SA node of the heart. T-type calcium channel blockers are used primarily as antiepileptics.10
L-type calcium channel
L type channel are particularly important in regulating contraction of cardiac muscle and smooth muscle (rang and dales). Long-lasting (L-type) voltage-gated calcium channels are found in both excitable and no excitable tissue.They are responsible for normal myocardial and vascular smooth muscle contractility. Five subunits (alpha-1, alpha-2, beta, gamma, and delta) make up the L-type channel. The alpha-1 subunit is the binding site for calcium-based antagonists. Dihydropyridine-based calcium antagonists are used as these binding sites.11
N-type calcium channel
N-types and P/Q-type of calcium channels play specialized roles in control neurotransmitter release (rang and dales). It has been known that the N-type calcium channel guards the synaptic transmission that conveys pain signals from sensory afferent nerve cells to the central nervous system. Certain naturally occurring peptide neurotoxins that specifically block N-type calcium channel have been shown to act as extremely potent and efficient analgesics in a wide range of animal pain models, including models of inflammatory and neuropathic pain. The available evidence suggests that N-type calcium channel blockers are at least as efficacious as opiates, are devoid of a number of the typical opiate side effects (e.g. respiratory depression) and that the analgesic effect is not subject to tolerance.
Table-2 Types and functions of voltage-gated calcium channel7
|
Type |
Gated by |
Characteristics |
Location |
Function |
|
L-type |
High Voltage |
High activation threshold, slow inactivation |
Plasma membrane of many calls. |
Main Ca+2 source for contraction in smooth and cardiac muscle |
|
N-type |
High voltage |
Low activation threshold, slow inactivation |
Throughout the brain |
Main Ca+2 sources for transmitter release by nerve terminals. |
|
P-type/ Q-type |
High voltage |
Low activation threshold, slow inactivation |
Nerve terminals |
Neurotransmitter Release
|
|
R-type |
Intermediate Voltage |
Low threshold, Fast inactivation |
Cerebellar granule cells, other neurons. |
?
|
|
T-type |
Low Voltage |
Low threshold, Fast inactivation |
Neurons, cells that have pacemaker activity. |
Regular Sinus Rhythm |
II. Ligand-gated channel
Ligand-gated cation channel that are activated by excitatory neurotransmitter are relatively non-selective, and conduct Ca+2 ions as well as other cations. Activation of receptor can readily cause so much Ca+2 entry that the cell dies, mainly through activation of Ca+2 –dependent proteases but also by triggering apoptosis.7
Table-3 Types and functions of Ligand-gatedcalcium channel
|
Type |
Gated by |
Characteristics |
Location |
Function |
|
IP3 receptor |
IP3 (Inositol trisphosphate) |
Ligand-gated channel activated by IP3 |
ER/SR |
Releases calcium from ER/SR in response to IP3 by e.g. GPCRs |
|
Ryanodine receptor |
Ca+2,sensitised by cyclic ADP ribose |
Directly activated in striated muscle via receptor of T tubules. |
ER/SR |
Calcium-induced calcium release in muscle |
|
Store-operated channels |
Indirectly by ER/SRdepletion of calcium |
Indirectly coupled to ER/SR Ca+2 stores. |
Plasma membrane |
Activated indirectly by that deplete intracellular stores e.g. GPCR agonists |
|
Two-pore channel |
------ |
------ |
------ |
------ |
|
Cation channels of sperm |
------ |
------ |
------ |
------ |
III. Store- operated calcium channel
Store- operated calcium channel are channel that occur in the plasma membrane and open to allow Ca+2 entry when the ER stores are decreases. They are distinct from other membrane calcium channel, and belong to large, recently discovered group of TRP (standing for transient receptor potential) channels, which have many different functions.
· Calcium extrusion mechanism
Active transport of calcium ions outwards across the plasma membrane, and inwards across the membrane of the ER or SR, depends on the activity of a Ca2+ -dependent ATPase. Several subtypes of the Ca2+-dependent ATPase have been cloned, but the physiological significance of this heterogeneity remains unclear. They have not been implicated in pharmacological responses, with the exception that thapsigargin specifically blocks the ER pump, causing loss of Ca2+ from the ER.
Calcium is also extruded from cells in exchange for Na+, by Na+- Ca2+ exchange. The exchanger transfers three Na+ ions for one Ca2+ ,and therefore produces a net depolarizing current when it is extruding Ca2+. The energy for Ca2+ extrusion comes from the electrochemical gradient for Na+, not directly from ATP hydrolysis. This means that a reduction in the Na+ concentration gradient resulting from Na+ entry will reduce Ca2+ extrusion by the exchanger, causing a secondary rise in free calcium ion , a mechanism that is particularly important in cardiac muscle. The exchanger can actually function in reverse if free sodium ion rises excessively, resulting in increased Ca2+ entry into cell.
· Calcium release mechanisms
There are two main types of calcium channel in the ER and SR membrane, which play an important part in controlling the release of Ca2+ from these stores.
The Inositol trisphosphate receptor (IP3R) is activated by Inositol trisphosphate (IP3), a second messenger produced by the action of many ligands on G-protein-coupled receptors .IP3R is a Ligand-gated ion channel, although its molecular structure differs from that of Ligand-gated channels in the plasma membrane. This is the main mechanism by which activation of G-protein-coupled receptors causes an increase in free calcium ions.
The Ryanodine receptor (RyR) is so called because it was first identified through the specific blocking action of the plant alkaloid Ryanodine. It is important in skeletal muscle, where there is direct coupling between the RyRs of the SR and the Dihydropyridine receptors of the T-tubules; these coupling results in Ca2+ release following the action potential in the muscle fibre. RyRs are also present in other types of cell that lack T tubules; they are activated by a small rise in free calcium ions , producing the effect known as calcium-induced calcium release (CICR), which serves to amplify the Ca2+ signal produced by other mechanisms such as opening of calcium channels in the plasma membrane. CICR means that release tends to be regenerative, because an initial puff of Ca2+ releases more, resulting in localised 'sparks' or 'waves' of Ca2+ release7.
The functions of IP3Rs and RyRs are modulated by a variety of other intracellular signals, which affect the magnitude and spatiotemporal patterning of Ca2+ signals.
A typical free calcium ions signal resulting from activation of a G-protein-coupled receptor. The response produced in the absence of extracellular Ca2+ represents release of intracellular Ca2+. The larger and more prolonged response when extracellular Ca2+ is present shows the contribution of SOC-mediated Ca2+ entry 7.
1.6 Calcium channel blockers
Calcium channel blockers (CCBs) are a heterogeneous group of drugs whose main pharmacological effect is to prevent or slow the entry of calcium into cell via specialized calcium channel14, 16. Calcium Channel blocker also called slow channel calcium antagonists, calcium antagonists, calcium blockers, slow channel blockers and calcium channel antagonists.15
Calcium channel blockers are drugs that slow the movement of calcium into the cells of the heart and blood vessels. Calcium channel blockers are group of drugs that reduced intracellular calcium transfer, thereby limiting calcium-triggered ATP release and thus reducing contractility of the muscle cell. Calcium channel blocker act especially on this tissue in which the slow calcium channel opens without being triggered by the fast sodium channel, e.g., the nodal tissue in the heart and the arterial smooth muscle.15
The calcium channel blockers inhibit the entrance of calcium into cardiac and smooth muscle cells of the coronary and systemic arterial. All calcium channel blockers are vasodilators that cause a decrease in smooth muscle tone and vascular resistance. These agents affect primarily the resistance of vascular smooth muscle and the myocardium.Calcium plays a critical role as an intracellular signal, and controls many different cell processes, of which calcium appears to play an important role in cell growth.17
Calcium channel blockers an important class of cardiovascular drugs. Members of the class are clinically useful in the treatment of cardiovascular disorders in which calcium plays a regulatory role16. Calcium channel blockers (CCBs) are a highly prescribed class of drugs used for the treatment of angina pectoris(chest pain), hypertension, and cardiac arrhythmias(irregular heartbeats)19.Other disorders include congestive heart failure, ischemic injury, and stroke, and migraine, tumour resistance to anti-neoplastic drugs, oesophageal spasms, bronchial asthma, and dysmenorrhoea which treat using calcium channel blocker16.
The main clinical usage of calcium channel blockers is to decrease blood pressure.10
Calcium channel blockers are useful in lowering blood pressure, controlling symptoms, and treating complications of a heart attack (such as arrhythmias). They may also be used if you cannot tolerate a beta-blocker.21
· History
Cardiovascular diseases are the first major disorder in world.Calcium channel blocker agents are some of the most widely prescribed drugs in America.First identified in the late 1960s, calcium channel blockers are non-habit-forming medications that are used to relax the smooth muscles of the arteries and arterioles as well as the heart muscle, which reduces the workload on the heart and causes a drop in blood pressure.25
Calcium channel blockers (CCBs) were at first introduced for use in the United States in 1981. Sustained-release formulations were available 10 years later.CCBs are frequently used during pregnancy for pregnancy-induced hypertension and to inhibit pre-term labor. These agents are helpful in the treatment of pre-term labor because they decrease myometrial contractility in the uterus. Generally, calcium channel blockers are administered in the second and third trimesters of pregnancy.34
Calcium channel blockers are most effective when they are combined with nitrates and beta-blockers, but their dosage must be monitored carefully to prevent side effects. The long-acting forms (taken once per day) of calcium channel blockers are preferred over the short-acting forms (taken 2 to 4 times per day).21
·Classes of calcium channel blockers Commercially available calcium channel blockers belong to one of three chemical classes:
1. Dihydropyridine Amlodipine, Aranidipine, Azelnidipine, Barnidipine, Benidipine,Cilnidipine, Clevidipine
2. Phenyl alkyl amines, Verapamil, Gallopumil
3. Benzothiazepine Diltiazem.3
How do calcium-channel blockers work?
Calcium-channel blockers slow the rate at which calcium passes into the heart muscle and into the vessel walls. This relaxes the vessels. The relaxed vessels let blood flow more easily through them, thereby lowering blood pressure. 27
Mechanism of action
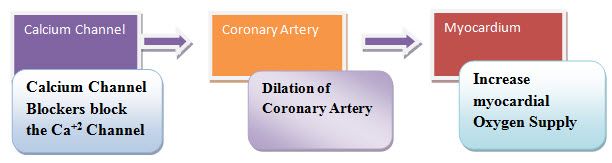
Fig.1. Mechanism of calcium channel (srspharma.com)
The intracellular concentration of calcium plays an important role in maintaining the tone of smooth-muscle and in the contraction of the myocardium.Calcium enters muscle cells through special voltage-sensitive calcium channels. Calcium channel antagonists block the inward movement of calcium by binding to L-type calcium channels in the heart and in smooth-muscle of the coronary and peripheral vasculature.14
Calcium channel blockers work by blocking voltage-gated calcium channels(VGCCs) in cardiac muscleand blood vessels.They prevent the calcium ions needed for muscle contraction from entering the cells of smooth and cardiac muscle. This decreases intracellular calcium leading to a reduction in muscle contraction. In the heart, a decrease in calcium available for each beat results in a decrease in cardiac contractility. In blood vessels, a decrease in calcium results in less contraction of the vascular smooth muscleand therefore an increase in arterial diameter, a phenomenon called vasodilatation.These vasodilating agents relax smooth muscle by inhibiting the entry of calcium into muscles or by inhibiting its release from intracellular storages.Vasodilatation decreases total peripheral resistance, while a decrease in cardiac contractility decreases cardiac output. Since blood pressure is determined by cardiac output and peripheral resistance, blood pressure drops. Calcium channel blockers result in a decrease in blood pressure, the bar receptor reflex often initiates a reflexive increase in sympathetic activity leading to increased heart rate and contractility. This causes the muscles to relax, lowering blood pressure, slowing the heart rate and decreasing oxygen demands of the hear10
· Pathophysiology of calcium-channel blockers
Calcium channel blockers have the following 4 cardiovascular effects:
- Peripheral vasodilatation (Smooth muscle relaxation)
- Negative chronotropy (decreased heart rate)
- Negative inotropy (decreased cardiac contractility)
- Negative dromotropy (decreased cardiac conduction).25
·General pharmacology Calcium channel blockers (CCBs) bind to L-type calcium channels located on the vascular smooth muscle, cardiac myocyte, and cardiac nodal tissue (sinoatrial and atrioventricular nodes). These channels are responsible for regulating the influx of calcium into muscle cells, which in turn stimulates smooth muscle contraction and cardiac myocyte contraction. In cardiac nodal tissue, L-type calcium channels play an important role in pacemaker currentsand in phase 0 of the action potentials. Therefore, by blocking calcium entry into the cell, CCBs cause vascular smooth muscle relaxation (vasodilatation), decreased myocardial force generation (negative inotropy), decreased heart rate (negative chronotropy), and decreased conduction velocity within the heart (negative dromotropy), manly at the atrioventricular node29
Side effects of calcium channel blocker
Calcium channel blockers have the following common side effects:
1. A slowed heart rate or irregular heart rhythm.
2. Flushing, dizziness, headache.
3. Leg and/or ankle swelling.
4. Decreased blood pressure (hypotension).
5. Tingling sensations in the arms or legs.
6. Weakness.
7. Constipation.
8. Skin rash.
9. Dryness of mouth.30
· Therapeutic applications
The calcium channel–blocking drugs have been investigated for an unusually wide number of clinical applications.
1. Hypertension
2. Angina pectoris
3. Arrhythmias.30
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
1.7 CALCIUM UPTAKE INHIBITING ACTIVITY39, 40
1.7.1 In vitro methods
1.7.1.1. 3H-nitrendipine binding in vitro
Procedure
The tissue was homogenized in ice cold buffer. 1 g cerebral wet weight/50 ml buffer, and centrifuged at 48,000 rpm(4°C) for 10 min. The resulting pellets are resuspended in approx. 270 ml buffer, and the homogenate was centrifuged at 48 000 g (4 °C) for 10 min. The final pellets are dissolved in buffer and 1 g cerebral cortex wet weight dissolved in 30 ml buffer. The membrane suspension is immediately stored in aliquots of 5–10 ml at –77 °C. Protein content of the membrane suspension is determined with bovine serum albumin as a standard. At the day of the experiment, the required volume of the membrane suspension is slowly and centrifuged at 48,000 rpm (4°C) for 10 min. The resulting pellets are resuspended in a volume of ice-cold incubation buffer, yielding a membrane suspension with a protein content of 0.6–0.8 mg/ml. After homogenization, the membrane suspension is stirred under cooling for 20–30 min until the start of the experiment. For each concentration samples are prepared in triplicate. The total volume of each incubation sample is 200 μl.
The binding reaction is started by adding 100 μl membrane suspension per incubation sample (0.6–0.8 mg protein/ml). The samples are incubated for 60 min in a bath shaker at 25 °C. The reaction is stopped by subjecting the total incubation volume to rapid vacuum filtration over glass fibre filters. Thereby the membrane bound is separated from the free radioactivity. Filters are washed immediately with approximately. 20 ml ice-cold rinse buffer per sample. The retained membrane-bound radioactivity on the filter is measured after addition of 2 ml liquid scintillation cocktail per sample in a Packard liquid scintillation counter.
The following parameters are calculated:
• Total binding
• Non-specific binding
• Specific binding = total binding – non-specific binding
1.7.2. Isolated organ38
1.7.2.1 Calcium antagonism on action potential of isolated guinea pig papillary muscle
Procedure
Guinea pigs of either sex weighing 300–400 g are sacrificed by stunning, the carotid arteries are severed, and the thoracic cage is opened immediately. The heart is removed, placed in a container of prewarmed, pre-oxygenated Ringer solution, and the pericardium and the atria are trimmed away. The left ventricle is opened and the two strongest papillary muscles removed. They are fixed between a suction electrode for electrical stimulation and a force transducer for registration of contractions. A standard micro electrode technique is applied to measure the action potential via a glass micro electrode containing 3 M KCl solution, which is inserted intracellular. The papillary muscle is stimulated with rectangular pulses of 1 V and of 1 ms duration at intervals of 500 ms. The interval between two stimuli is variable in order to determine refractory periods. The intensity of the electrical current is just below the stimulation threshold. The intracellular action potential is amplified, differentiated for registration of upstroke velocity, together with the contraction force displayed on an oscilloscope, and recorded. After an incubation period of 30 min the Ringer solution was changed. For further depolarization, isoproterenol (1.0 mg per 100 ml) is added. By this measure, resting potential is increased to about 40 mV, resulting in inactivation of the fast inward sodium channel. The resulting slow rising action potential is sensitive to calcium antagonistic drugs. The test compound is added at a concentration of 1 μg/ml. Effective compounds are tested at lower concentrations and compared with the standard Nifedipine at concentrations of 0.01 and 0.1 μg/ml.
1.7.2.2. Calcium antagonism in the isolated guinea pig






