

 ABOUT AUTHORS:
ABOUT AUTHORS:
1 Shashi Kant*,Satinder Kumar, Rajender Kumar,
Research scholar department of pharmacy.
2 Dr. Bharat Prashar
HOD & Associate professor Department of Pharmaceutical Sciences, Manav Bharti University, Solan (H.P)
* shashi_ranaute@yahoo.in
ABSTRACT:-
In this review article, we discussed about Bioavailability and bioequilance study, bioavailability means rate and extent to which active ingredients absorbed from a drug product and become available at site of action. This article provides the information about important aspect involved in bioequivalence and regulatory requirement for bioequivalence study. The rate or rapidity at which drug is absorbed is important consideration in treatment of acute conditions such as asthma attack in pain. Extent of absorption is of special significance in treatment of chronic conditions like hypertension, epilepsy. In this review we discussed all the factors affecting bioavailability from its dosage form, objective/purpose of bioavailability study, design and evaluation of bioequilance study, methods of assessing of bioavailability and bioequilance etc.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-1326
INTRODUCTION:-
Bioavailability means rate and extent to which active ingredients absorbed from a drug product and become available at site of action.
The rate or rapidity at which drug is absorbed is important consideration in treatment of acute conditions such as asthma attack in pain. Extent of absorption is of special significance in treatment of chronic conditions like hypertension, epilepsy etc.
There are some definitions from 2003 orange book, code of federal regulations, 21 CFR 320 and other source:-
Pharmaceutical alterative: drug products that contain same therapeutic moiety but as different salts, esters or complexes. For example tetracycline phosphate or tetracycline hydrochloride equivalent to 250mg tetracycline base are considered pharmaceutical alternative.
Pharmaceutical equivalent: drug product in identical dosage form that contain same active ingredients that is same salt or ester, are of same dosage form, use same route of consideration, identical in strength.
Pharmaceutical substitution: the process of dispensing a pharmaceutical alternative for prescribed refers to comparison of bioavailability of different formulations, drug product or batches of same drug product. drug product. Eg. Ampicillin suspension is dispensed in place of Ampicillin capsule.
Therapeutic alternative: drug product containing different active ingredients that are indicated for same therapeutic or clinical objective. Eg Ibuprofen is given instead of aspirin.
Therapeutic substitution: process of dispensing a therapeutic alternative in place of prescribed drug product eg amoxicillin is dispensed instead of ampicillin.[1][2][3][4]
FACTORS AFFECTING BIOAVAILABILITY OF DRUGS FROM ITS DOSAGE FORM
[adsense:468x15:2204050025]
A. DRUG SUBSTANCE PHYSICOCHEMICAL PROPERTIES
1. Particle size
2. Crystalline or amorphous form
3. Salt form
4. Hydration
5. Lipid or water solubility
6. PH and pKa
7. Dissolution rate and drug solubility
B. PHARMMACEUTICAL INGREDIENTS
1. Fillers
2. Binders
3. Coating
4. Disintegrating agent
5. Lubricants
6. Suspending agent
7. Surface active agent
8. Flavoring agent
9. Coloring agent
10. Preservative
11. Stabilizing agent
C. DOSAGE BFORM CHARECTERISTICS
1. Disintegration rate
2. Dissolution time of drug in dosage form
3. Product age and storage condition
D. PHYSIOLOGICAL FACTORS AND PATIENT CHARECTERISTICS
1. Gastric emptying time
2. Intestinal transit time
3. Pathological condition(gastrointestinal abnormality)
4. Gastric contents
5. Food
6. Gastrointestinal PH
E. ROUTE OF ADMINISTRATION
The influence of route of administration on drug bioavailability is generally following order: Parenteral > oral > rectal > topical
Within parenteral route, intravenous injection of drug results in 100% bioavailability.
The amount of drug that reaches systemic circulation called systemic bioavailability.
Bioavailable fraction (F) refers to fraction of administered dose that enters systemic circulation.
F = Bioavailable dose/ Administered dose
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
OBJECTIVE/ PURPOSE OF BIOAVAILABILITY STUDIES
1. Primary stage for development of suitable dosage form for new drug entity.
2. Determination of influence of excipients, patient related factors and possible interactions with other drugs on efficiency of absorption.
3. Development of new formulation of existing drug
4. Control of quality of drug product during early stages of marketing in order to determine influence processing factors, storage and stability on drug absorption.
TYPES OF BIOAVAILABILITY
- Absolute bioavailability
- Relative bioavailability/Comparative bioavailability
ABSOLUTE BIOAVAILABILITY:
It is systemic availability of drug after extra vascular administration (eg.oral, rectal, transdermal) compared to IV dosing. It is denoted by F. IV dose is selected as standard if drug is poorly water soluble. There are several drawbacks of using oral solution as a standard instead of IV dose:
1. Limits pharmacokinetic treatment to one-compartment model only.
2. Differentiation between fraction of dose unabsorbed and that metabolized is difficult.
Absolute BA = (AUC)PO/(Dose)PO
(AUC)IV / (Dose)IV
Absolute BA using urinary excretion data can be determined as follows;
Absolute BA = (Du)po∞/(Dose)PO
(Du)IV∞/(Dose)IV
For drug given intravascularly (IV), F = 1
For all extra vascular route of administration, such as oral route, F>1
Absolute bioavailability (F) expressed in Percentage.
RELATIVE BIOAVAILABILITY:
Also called as comparative bioavailability. Defined as availability of drug from drug product as compared to reference standard. The relative BA of 2 drug products given at same dosage level and by same route of administration can be obtained by following equation.
Relative availability (Fr) = (AUC)A/ (AUC)B
Where drug product B is reference standard.
When different dose are administered, then
Relative availability (Fr) = (AUC)A/(Dose)A
(AUC)B/ (AUC)B
The percent relative availability using urinary excretion data can be determined as follows;
Percent relative availability = (DU)A∞ ×100
(DU)B∞
Where (DU)∞ = total amount of drug excreted in urine.
Fr also expressed in percentage.
HUMAN VOLUNTEERS – HEALTHY SUBJECTS VERSUS PATIENTS
Ideally BA studies should be carried out in patients;
Advantages:
(1) reflects better therapeutic efficacy of drug
(2) Drug absorption pattern in disease state can be evaluated.
Disadvantage: patients are generally preferred in multiple dose studies. Drawback is diseases state may modify absorption pattern.
Bioavailability studies usually performed in young (20-40yrs), healthy male adult volunteers under restricted diet. Medical examination should be performed in order to exclude subjects with any kind of disease.
METHODS FOR ASSESSING BA/BE:
Direct and indirect methods are used to asses drug BA. The in vivo demonstrated by comparison of measured parameters eg. Concentration of active ingredients in blood, cumulative urinary excretion rate etc.
Pharmacokinetic and pharmacodynamic parameters as well as clinical observation and in vitro studies used to determine drug BA.
A. Pharmacokinetic method
1. Plasma drug concentration:
Time for peak plasma concentration (tmax)
Peak plasma drug concentration (Cmax)
Area under plasma drug concentration time curve (AUC)
2. Urinary drug excretion
Cumulative amount of drug excreted in urine (Du)
Rate of drug excretion in urine (dDu/dt)
Time for maximum urinary excretion (t)
B. Pharmacodynamic method
1. Acute pharmacological response
2. Therapeutic response
C. In vitro studies
Drug dissolution
PLASMA DRUG CONCENTRATION:
It is most reliable method.
With single dose studies, method requires collection of serial blood sample for period of 2-3 biological half lives after drug administration.
With multiple dose studies method involve drug administration for at least 5 half-life.
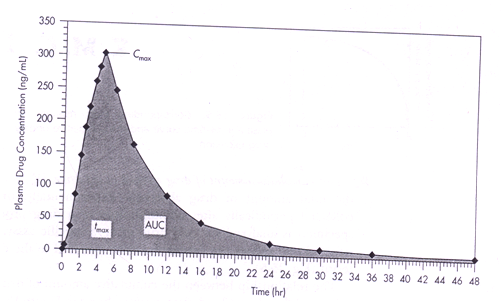
1. tmax :
The time of peak plasma concentration, corresponds to time required to reach maximum drug concentration after drug administration. At tmax peak drug absorption occur and rate of drug absorption equals rate of drug elimination. Drug absorption still continue after tmax is reached, but at slow rate.
Plasma Drug Conc - time curve
tmax used as an approximate indication of drug absorption rate.
Units: hours, minutes
2. Cmax:
Peak plasma drug concentration, represents maximum plasma drug concentration obtained after oral administration.
It provides indication that drug is sufficiently systemically absorbed to provide therapeutic response. It also provides warning of possibly toxic level of drug.
Units: mg/ml, ng/ml
3. AUC:
Area under plasma level time curve is measurement of extent of drug bioavailability. The AUC reflects total amount of active drug that reaches systemic circulation.
The AUC from t=0 to t=∞ is equal to amount of unchanged drug reaching general circulation divided by clearance.

F = fraction of dose absorbed, DO = Dose
VD = volume of distribution, K = Elimination rate constant
The AUC independent of route of administration
Units: µg.hr/ml
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Many methods are available for measuring AUC.
1. CUT AND WEIGH METHOD: in this method plasma concentration profile are plotted on smooth texture paper, these can be cut out and weighed on an electronic balance. The weight of these cut out plot will be proportional to AUC.
2. Planimeter is also used in calculating AUC. It is an precise instrument which allows calculation of areas by tracing their outerlines.
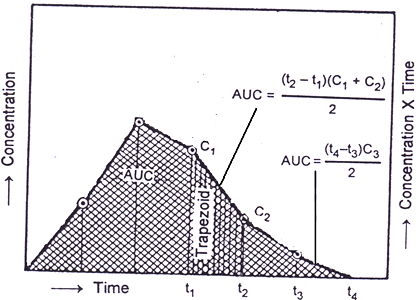
3. Trapezoidal method: It is simplest method and involves breaking up of plasma concentration versus time profile into several trapezoids; calculate area of individual trapezoids and then adding up these areas to arrive at cumulative AUC.
AUC = (CO + C1)(t1 – t0)/2 + (C1+C2)(t2-t1)/2 + -------------+ (Cn-1+Cn)(tn-tn-1)/2
Integration method:
The rate of change of plasma concentration(C) is described as
C = A (e-Kt – e-Kat)
Dc/dt = rate of absorption – rate of elimination
= Ka Xa – KX
Where ka and K absorption and elimination rate constant
Xa and X are amount of drug in GIT and body respectively
An integration of this equation between limits of time for which the drug remain in body, as reflected by plasma concentration, gives
And the total area under curve (AUC), for which total integral time zero and infinity is give by
AUC = A (1/K – 1/Ka)
Or simply AUC from t=0 to t=∞, equal to amount of unchanged drug reaching general circulation divided by clearance.
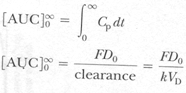
Where F = fraction of dose absorbed, D = Dose
K = elimination rate constant, VD = Volume of distribution
URINARY DRUG EXCRETION DATA:
It is an indirect method of estimating BA. Timely drug sample must be collected and total amount of urinary drug excretion must be obtained.
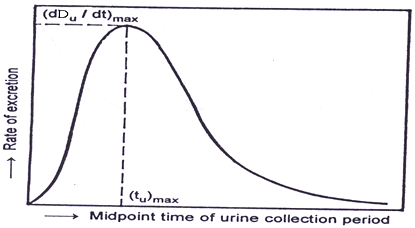
1. Du ∞: Is Cumulative amount of drug excreted in urine related directly to total amount of drug absorbed.
Experimentally urine sample are collected periodically after drug administration. Each urine specimen is analyzed for free drug urine specific assay.
The graph is constructed that relates cumulative amount of drug excreted to collection time interval.
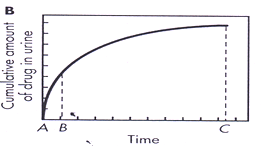
2. dDu / dt:
Rate of drug excretion, It depend on first order elimination rate constant K and concentration of drug in plasma CP.
Figure shows maximum rate of drug excretion is at point B. whereas minimum rate of drug absorption is at point A and C.
3. t∞ : Total time for drug to be excreted.
Its value decreases as absorption rate increases.
t∞ is useful parameter in bioequivalence studies that compare several drug products.
PHARMACODYNAMIC METHODS:
1. Acute pharmacological response:
When bioavailability measurement by pharmacokinetic methods is difficult, inaccurate or nonreproducilble, an acute pharmacological effect such as change in ECG or EEG reading, pupil diameter, BP is related to time course of given drug.
Bioavailability can be determined by construction of pharmacological effect time curve as dose response graph.
The method requires measurement of at least three biological half life of drug in order to obtain good estimate of AUC.
Disadvantage of this method is that pharmacological response is more variable and accurate correlation is difficult.
2. Therapeutic response:
In this method clinical response of drug for which it is to be intended to be used is measured. eg. For anti-inflammatory drugs, reduction in inflantation is determined.[3][4]
BIOEQUIVALENCE STUDIES:
The in vivo BE study requires determination of relative BA after administration of single dose of test and reference formulation by same route, in equal dose but at different times. The reference product is generally approved product, usually innovators product.
DESIGN AND EVALUATION OF BIOEQUIVALENCE STUDIES
Bioequivalence studies are performed to compare the BA of generic drug product to brand name product statistical techniques should be of sufficient sensitivity to detect difference in rate and extent of absorption.
The design and evaluation requires co-operative inputs from pharmacokinetics, statisticians, clinicians, bioanalytical chemists and others. The basic design for BE study is determined by;
1. Scientific questions to be answered
2. Nature of analytical methods
3. Availability of analytical methods
4. Benefits-risk and ethical consideration with regard to testing in human
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
ELEMENTS OF BA STUDY PROTOCOL
|
1. Title 2. Study objective 3. Study design Title a. Drug product Test product Reference product b. Dosage regimen c. Sample collection schedule d. Fasting/meal schedule e. Analytical method 4. Study population a. Subjective b. Subject selection Medical history Physical examination |
5. Clinical procedures a. Dosage and drug administration b. Biological sampling schedule c. Activity of subject 6. Ethical consideration a. Basic principle b. Informed consent c. Indication for subject withdrawal d. Adverse reaction and emergency procedure 7. Facilities 8. Data analysis 9. Drug accountability 10. Appendix |
ANALYTICAL METHODS:
The analytical method used in BA or BE studies to measure concentration of active drug must be accurate and sufficient, with appropriate precision. For BA studies, both parent drug and its major active metabolites are measured. For BE studies, the parent drug is measured.
REFERENCE STANDARD:
For BE studies, reference standard is Reference Listed Drug (RLD), which is listed in approved drug product with therapeutic equivalence evaluation-the orange book. The RLD is generally a formulation currently marketed with fully approved NDA for which there are valid scientific safety and efficacy data. The RLD is usually innovators or original manufacturer’s brand name product.
STUDY DESIGN:
Currently 3 different studies may be required for solid oral dosage form including;
1. Fasting study
2. Food intervention study
3. Multiple dose study
Proper study design and statistical evolution are important consideration for determination of BE.
FASTING STUDY:
This study is required for all immediate release and modified release dosage form. Both male and female are used in study. Blood sampling is performed just before the dose and at appropriate interval after dose to obtain adequate description of plasma drug concentration time profile. The subject should be in fasted state before drug administration and should continue to fast up to 4hr after design. No other medication is normally given to subject at least 1 week prior to study.
FOOD INTERVENTION STUDY:
Co-administration of food with oral drug product may affect bioavailability of drug. Food intervention studies are generally conducted using meal condition. The test meal is high fat and high calorie meal. A typical test meal 2 eggs fried in butter, 2 strips of bacon, 2 slices of toast with butter, 4 ounces of brown potatoes and 8 ounces of rice. Foe BE studies, subjects are given recommended meal 30 mint before design.
MULTIPLE DOSE STUDY: In few cases, multiple dose study comparing equal dose of test and reference product may be performed in adult, healthy subjects.
CROSS OVER DESIGN:
1. LATIN SQUARE CROSS OVER DESIGN:
In which each formulation is administered just once to each subject and once in each study period. Unlike parallel design, all subjects do not receive same formulation in same time, in a given study period, they are administered different formulation.
Advantage:
It minimize intersubject variability in plasma level
Minimize variation due to time effect
Minimize intra-subject variability
Drawback: Study takes long time
When number of formulation to be tested are more, study become more difficult
|
Subject
1 2 3 4 5 6 |
Study period 1
A B C A C B |
Study period 2
B C A C B A |
Study period 3
C A B B A C |
2. REPLICATED CROSS OVER DESIGN:
In this same reference and same test are given to same subject. In this design, reference to reference and test to test comparison may also be made. Generally a four period, 2 sequences, 2 formulation designs are recommended by FDA.
|
Sequence 1
Sequence 2
|
Period 1
T
R |
Period 2
R
T |
Period 3
T
R |
Period 4
R
T |
Where R = reference and T = treatment
EVALUATION OF DATA:
1. ANALYTICAL METHOD:
The analytical method for measurement of drug must be validated for accuracy, precision, sensitivity and specificity. The use of more than one analytical method during BE studies may not be valid because different methods may yield different values.
2. PHARMACOKINETIC EVALUATION OF DATA:
For single dose studies, including fasting study or food interaction study, pharmacokinetic analysis include calculation of each subject of (AUC0→t) and to infinity (AUC0→∞), tmax and Cmax. Additionally, elimination rate constant K and elimination half life (t1/2) may be estimated.
For multiple dose studies, analysis include calculation for each subject of (AUC0→t), tmax , Cmin, Cmax and percent fluctuation.
STATISTICAL INTERPRETATION OF BIOEQUIVALENCE DATA:
After data has been calculated, statistical methods must be applied to determine level of significance in order to establish BE between two or more drug product.
Typically ANOVA analysis of variance method is applied to determine statistical difference. Currently a simple acceptable rule is that if related BA of test formulation is in range of 80-120% of reference product, it is considered bioequivalent.
ANOVA may evaluate variability in subjects, treatment ground, study period, formulation and other variables depending on study design.[5-11]
REFERENCES:
- Applied Biopharmaceutics and Pharmacokinetics-5th edition by Leon Shargel, Susanna Wu-PONG, Andrew B.C.YU. Page no; 453-498[1]
- Bioavailability and Bioequivalence in Pharmaceutical Technology by Tapan Kumar Pal, M.Ganesan; Page no: 9-29 [2]
- Biopharmaceutics and pharmacokinetics a treatise by D.M.Brahmankar, Sunil B. Jaiswal; Page no: 282-305 [3]
- Biopharmaceutics and Pharmacokinetics by G.R.Chatwal; Page no: 189-200 [4]
- Textbook of Biopharmaceutics and Pharmacokinetics by Sarfaraz Niazi; Page no: 61-62 [5]
- MHW guideline for bioequivalent studies of generic products, Dec 1997.[6]
- Saudi food and drug administration drug authority sector Bioequivalence requirments guidelines, (2005).[7]
- Stastical Approaches to Establish Bioequivalence US Food and drug administration center for evaluation and drug research FDA, (2001).[8]
- The European agency for the evaluation of medicinal products-CPMP/EWP/QWP/1401/98 Rev. 1/ Corr*[9]
- fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070111.pdf, [10]
- fda.gov/OHRMS/DOCKETS/98fr/3657gd2.pdf, [11]
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






