

{ DOWNLOAD AS PDF }
About Authors:
Yogeshkumar B. Viradiya*, Manoj B. Dagwar, Swapnil T. Lanjewar
Institute of Management Sciences and Research (IMSR),
Nagpur, Maharashtra
viradiya2210@gmail.com
Abstract:
European Union is the big market for pharmaceuticals. Every pharmaceutical company wants to starts their business in European Union. In European Union come different countries like UK, Germany, France, Ireland, Sweden etc. In these countries various different process for new drug registration process like Nationalize process, Centralize process, Decentralize process, Mutual recognition process. Before that clinical trial approval also important for new drug registration. New drug registration process in European Union takes approximately 33 to 35 weeks. Each company must follow the rules and regulation for new drug registration. European medicine agency which gives the Clinical trial authorization and Marketing authorization to the new drug.
Introduction
Clinical Trial Authorization in European Union.1
The application process to perform a clinical trial in Europe takes place on a country-by-country basis each country have a different process. Thus, a sponsor must apply for approval in each country in which it intends to have study sites. The processes are same in most countries, there are minor differences and, in some cases, additional material must be submitted. For example, in many European countries, a sponsor must submit a copy of the insurance coverage obtained to cover the clinical trial study.
In this document, we present the process which is used in the UK as it is a recognized standard.1
The regulatory authority that governs therapeutics in the UK is the Medicines and Healthcare products Regulatory Agency (MHRA). To file a clinical trial application in the UK, a sponsor must reside or have a legal representative in the EU.1
Major parts of a clinical trial application (CTA)1
A clinical trial application consists of several major parts:
• Covering letter: This should contain EudraCT (European Clinical Trials Database) number, the title and number of the study protocol, and information on any special issues such as first-time use in humans, use of special populations, or unusual trial design.1
• Clinical trial application form: The form is available from the website of EudraCT.
• Protocol: The protocol should include an evaluation of the anticipated benefits and risks, a justification for including any subjects who may not be able to provide informed consent (if applicable), and a description of the plan to provide additional care once patients leave the study, if different from normal medical care.1
• Investigator’s Brochure (IB) or Summary of Product Characteristics* (SmPC): The IB should be prepared from all available information and evidence that supports the rationale for the proposed clinical trial and safe use of the investigational product.1
• Investigational Medicinal Product-related data: Include the Investigational Medicinal Product Dossier (IMPD). This contains summaries of information related to the quality, manufacture and control of the investigational product. It should include chemical, pharmaceutical and biological data, non-clinical pharmacology and toxicology data, previous clinical trial and human experience data, and overall risk and benefit assessment. In cases where the investigational product has a marketing authorization in another EU member state or it has been approved in another pharmaceutical form, the sponsor can provide an abbreviated IMPD.1
• XML file of the application form: Provide the complete data set.
Applicable fee.1
Procedure for clinical trial approval 4
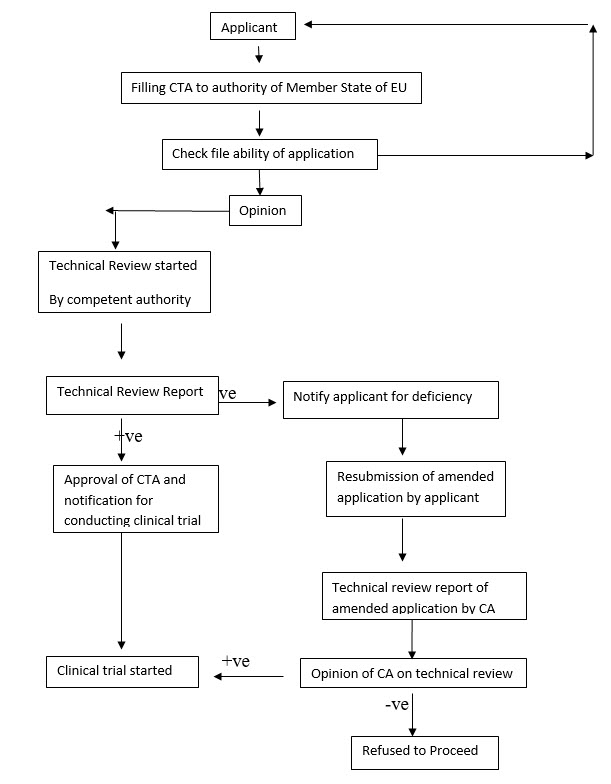
Figure 1: clinical trial authorization process in EU.
A guidance document published by the European Commission which gives the specific requirements for each country in the EU.
A sponsor is required to obtain a EudraCT number. EudraCT is a database of all clinical trials occurring in Europe. It is operated by the European Medicines Agency (EMA). A sponsor obtains a EudraCT number by accessing the site and completing a EudraCT application. For a clinical trial to be performed in multiple EU countries, only one EudraCT number is required.1
After submission of the CTA to the MHRA, the sponsor will receive an acknowledgement letter. If the CTA is judged as valid (i.e., all required information has been provided), the thirty-day assessment period begins. If the CTA is not valid, the sponsor will be informed of the deficiencies and be given an opportunity to update the file. If the deficiencies are major, the sponsor may be requested to withdraw the file and resubmit once it is complete.
Phase I studies involving healthy volunteers have a fourteen-day assessment period.1
Possible outcomes1
There are three potential outcomes after the review of the CTA:
1. Acceptance of the request for a clinical trial authorization.
2. Acceptance of the request for a clinical trial authorization subject to conditions.
3. Grounds for non-acceptance (GNA) of the request for a clinical trial authorization.
Grounds for Non-Acceptance letters1
The Grounds for Non-Acceptance (GNA) letter will identify the deficiencies or inadequacies in the submission. A sponsor has the opportunity to make an amended request for a CTA by addressing all the issues identified in the GNA letter. No additional changes are permitted. The possible outcomes from the sponsor’s amendment are similar to the three original potential outcomes:
1. Acceptance of the amended request.
2. Acceptance of the amended request subject to conditions.
3. Grounds for non-acceptance of the amended request.
The MHRA targets 60 days from the date of the original application to provide a response on the amendment to the sponsor. Phase I studies involving healthy adults will be assessed within 14 days. If a sponsor opts not to respond to a GNA letter, the application will be deemed to have been refused as of 60 days from the date of receipt of the original application. In this case, the MHRA does not send a notification letter.1
Initial Application1
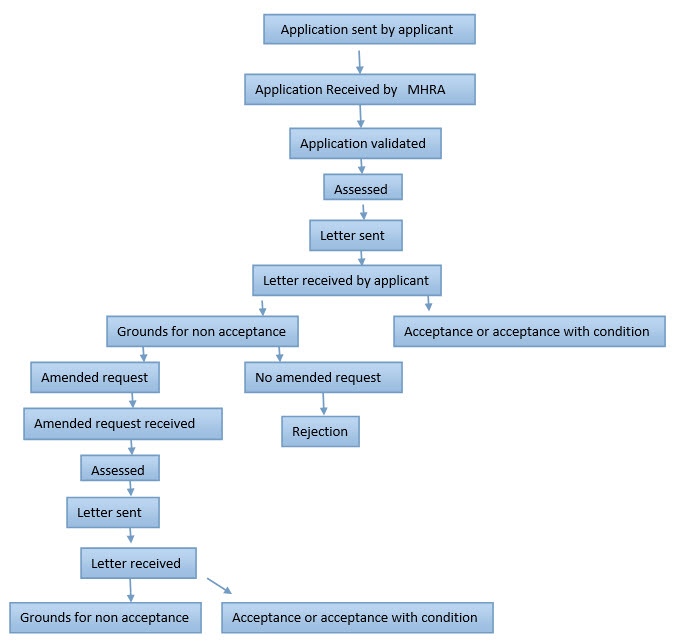
A clinical trial is not permitted to start until each site has received a favorable opinion from its Ethics Committee.1
Amendments to a trial1
The guidance document published by the European Commissionalso provides details on what types of amendments need to be submitted to regulators.
Non-substantial amendments do not have to be submitted to regulators. However, the changes should be recorded and available on request. In addition, they should be included in the next update to the Investigator’s Brochure, if appropriate.
Substantial amendments are those considered likely to have a significant impact on the:
• Safety or physical or mental integrity of the subjects
• Scientific value of the trial
• Conduct or management of the trial
• Quality or safety of any investigational product used in the trial
Substantial amendments are required to be submitted to each site’s Ethics Committee and the country-specific regulator. An amendment may be implemented once approval has been obtained from the Ethics Committee and the amendment has been submitted to the regulator and there are no grounds for non-acceptance.1
Safety reporting1
Similar to the requirements in the US and Canada, sponsors are required to submit safety information on unexpected drug-related serious adverse events to regulators in an expedited manner.
The MHRA must be notified of fatal or life-threatening suspected (meaning, drug-related) unexpected serious adverse events (SUSAR) in an expedited fashion and no later than seven calendar days after information on the SUSAR is received. The sponsor then has a further eight days to submit additional relevant information.
For non-fatal or non-life-threatening suspected unexpected serious adverse events, the report must be submitted within 15 calendar days after the information is received.1
Declaration of end of trial1
The end of a clinical trial is considered to take place when the last patient has the last visit. Exceptions to this definition should be justified in the protocol. The sponsor is required to notify the regulator once a trial has ended in the UK (or the given country) as well as in all other countries (if the trial is multinational in scope).
If a trial ends prematurely, the sponsor must notify the regulator within 15 days of when the trial is halted and explain the reasons for terminating the study. If a trial is temporarily halted, the sponsor must also notify regulators within 15 days. Restarting a halted trial requires submitting an amendment as it is considered to be a substantial amendment. If a sponsor elects not to restart a temporarily halted trial, it has 15 days to notify the regulator.1
Drug Approval Process in Europe union
Regulatory Agency Of European Union
European medicine agency (EMA)
European directorate for the quality of medicine (EDQM)
Types of process
Nationalized process
Decentralized process
Mutual recognition process
Centralize process
In European Union (EU), all European countries which are come in the European Union. There are no specific rules & regulation for different countries, same rules & regulation follow by all European countries for pharmaceutical. New drug registration process is same for all countries. All medical products were approved for marketing at the National level. The first reorganization procedure was introduced in 1983 and a single national review in case of pharmaceutical/medicinal product for marketing authorizations in all EU's countries was made feasible. The single aim of this procedure was to create a united standard for product review among national regulatory authorities.
For high-technology or biologically derived products, the concentration procedure was established by directive 87/22, in which product assessment should be completed by Committee for Proprietary Medicinal Products (CPMP) besides the normal national regulatory review it was possible in 1987. Further, in 1993, by council regulation (EEC) 2309/93, the concentration procedure was replaced with centralized procedure, by which all the high-tech and biologically derived product was reviewed and granted EU's wide marketing authorization by the EU's CPMP.
Two main phases for approval of new drug in European countries. Clinical trial and marketing authorization. A clinical trial application (CTA) is filed to the competent authority of the state to conduct the clinical trial within European Union. The competent authority of that member state evaluates and check the application. The clinical trials are conducted only after the approval of new drug. In the EU's countries, the companies have a choice of following regulatory procedures:
A sponsor has several options when seeking approval to market a new drug in Europe: a national authorization procedure, a decentralized procedure, a mutual recognition procedure, or a centralized procedure. Products that must use the centralized procedure include the following:5
• All biologic agents or other products made using high-technology procedures5
• Products for HIV/AIDS, cancer, diabetes, neurodegenerative diseases, auto-immune and other immune dysfunctions and viral diseases5
• Products for orphan conditions5
This report focuses on the centralized procedure, with a brief overview of the other three procedures provided below.5
National authorization procedure5
Each country within the EU has its own procedures for authorizing a marketing application for a new drug. A sponsor can consult the website of the regulatory agency in each country in which it is interested in obtaining marketing approval to obtain details of the approval process.
A sponsor can also seek approval of several EU countries simultaneously using the decentralized or mutual recognition procedure.5
The Centralized Procedure3
The 'centralized procedure' for authorizing medicinal products is laid down in Regulation (EC) No 726/2004.
The centralized procedure, which came into operation in 1995, allows applicants to obtain a marketing authorization that is valid throughout the EU. It is compulsory for medicinal products manufactured using biotechnological processes, for orphan medicinal products and for human products containing a new active substance which was not authorized in the Community before 20 May 2004 (date of entry into force of Regulation (EC) No 726/2004) and which are intended for the treatment of AIDS, cancer, neurodegenerative disorder or diabetes. The centralized procedure is also mandatory for veterinary medicinal products intended primarily for use as performance enhancers in order to promote growth of treated animals or to increase yields from treated animals.3
The centralized procedure is optional for any other products containing new active substances which is not authorized in the Community before 20 May 2004 or for products which constitute a significant therapeutic, scientific or technical innovation or for which a Community authorization is in the interests of patients or animal health at Community level.3
When a company wishes to place on the market a medicinal product that is eligible for the centralized procedure, it sends an application directly to the European Medicines Agency, to be assessed by the Committee for Medicinal Products for Human Use (CHMP) or the Committee for Medicinal Products for Veterinary Use (CVMP). The procedure results in a Commission decision, which is valid in all EU Member States. Centrally-authorized products may be marketed in all Member States.3
Applications from persons or companies seeking 'orphan medicinal product designation' for products they intend to develop for the diagnosis, prevention or treatment of life-threatening or very serious conditions that affect not more than 5 in 10,000 persons in the European Union are reviewed by the Committee for Orphan Medicinal Products (COMP). The Committee on Herbal Medicinal Products (HMPC) is responsible for establishing a list of herbal substances, preparations and combinations thereof for use in traditional herbal medicinal products. It is also responsible for establishing Community herbal monographs.3
Decentralized procedure5
For products that fall outside the scope of the European Medicines Agency (EMA [www.ema.europa.eu]) with regard to centralized procedures, a sponsor can submit under the decentralized procedure. Using this process, a sponsor can apply for simultaneous authorization in more than one EU country for products that have not yet been authorized in any EU country.5
Mutual recognition procedure5
With the mutual recognition procedure, a product is first authorized by one country in the EU in accordance with the national procedures of that country. Later, further marketing authorizations can be sought from other EU countries, who, rather than conducting their own review, agree to recognize the decision of the first country.5
Exhibit 1: Mutual recognition procedure5
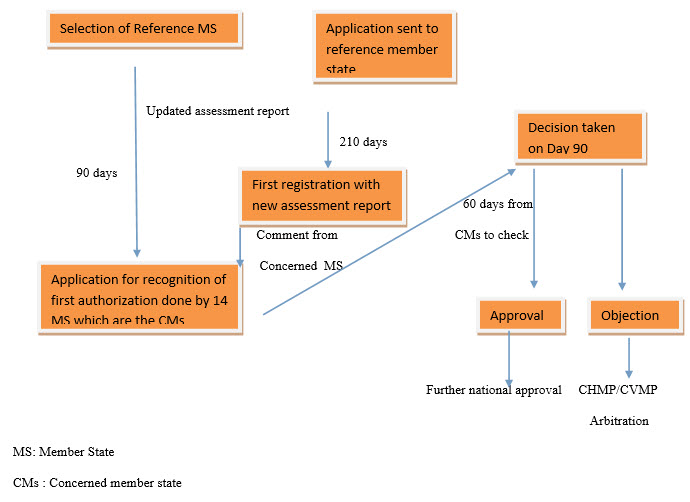
Exhibit 2: centralized procedure5
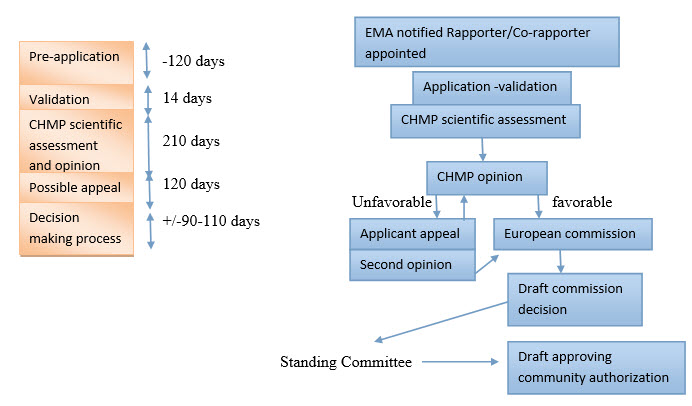
Exhibit 3: Marketing authorization application modules5
|
Module ( Detail) |
Content |
|
1 |
EU administrative and prescribing information *Application form *Summary of product characteristics *Labeling text and mock-ups *Information about the experts *Environmental risk assessment *Information relating to orphan-market exclusivity *Description of the Pharmacovigilance system *Risk management plan |
|
2 |
High-level summaries *Quality *Non-clinical overview *Non-clinical summary (pharmacology, pharmacokinetics, toxicology) *Clinical overview *Clinical summary (biopharmaceutics, clinical pharmacology, efficacy, safety, study synopsis) |
|
3 |
Quality documentation *Body of data *References Non-clinical documentation *Study reports *References |
|
4 |
Clinical documentation *Tabular listing of studies *Study reports *References |
Exhibit 4: Standard timetable for review of a centralized application5
Standard timetable for the evaluation of a centralized application
|
Day |
Action |
|
1 |
Start of the procedure. |
|
80 |
Receipt of assessment report or critique from reporter and co-reporter by CHMP members and EMA. EMA sends assessment report to applicant making clear it only sets out preliminary conclusions and is sent for information only and does not yet represent the position of the CHMP. |
|
100 |
Rapporteur, co-rapporteur, other CHMP members and EMA receive comments from CHMP members, including peer reviewers. |
|
115 |
Receipt of draft list of questions (including CHMP recommendation and scientific discussion) from rapporteur and co-rapporteur, as discussed with peer reviewers, by CHMP members and EMA. |
|
120 |
CHMP adopts list of questions as well as overall conclusions of scientific data to be sent to applicant by CHMP. Clock stops on timetable. |
|
121* |
Submission of the responses, including revised summary of product characteristics, labelling, and package leaflet texts in English, restart of clock. After receipt of responses: |
|
150 |
Joint response (assessment report) from rapporteur and co-rapporteur received by CHMP members and EMA. EMA sends joint-assessment report to applicant making clear it only has preliminary conclusions and does not yet represent position of CHMP. |
|
170 |
Deadline for comments from CHMP members to be sent to rapporteur and co-rapporteur, EMA and other CHMP members. |
|
180 |
CHMP discussion and decision on the need for adoption of a list of “outstanding issues” and/or an oral explanation by applicant. If oral explanation needed, clock is stopped to allow time for preparation. |
|
181 |
Restart clock and oral explanation (if needed). |
|
181-210 |
Final draft of English summary of product characteristics,labeling, and package leaflet sent by applicant to rapporteur and co-rapporteur, EMA and other CHMP members. |
|
By 210 |
Adoption of CHMP Opinion & CHMP Assessment Report (and timetable for provision of product information translations). After adoption of CHMP opinion |
|
215 |
at the Latest Applicant provides EMA with summary of product characteristics, labeling, and package leaflet in 20 Languages. EMA circulates draft to member states for review. |
|
232 |
at the latest Applicant provides EMA with final translations of summary of product characteristics, labeling, and package leaflet in 20 languages, taking into account comment received from member states by day |
|
By 237 |
Transmission of opinion and annexes in all EU languages to applicant, commission, members of standing committee, Norway and Iceland. |
|
By 246 |
Applicant provides EMA with one final full-color mock-up of outer and inner packaging for each pharmaceutical form |
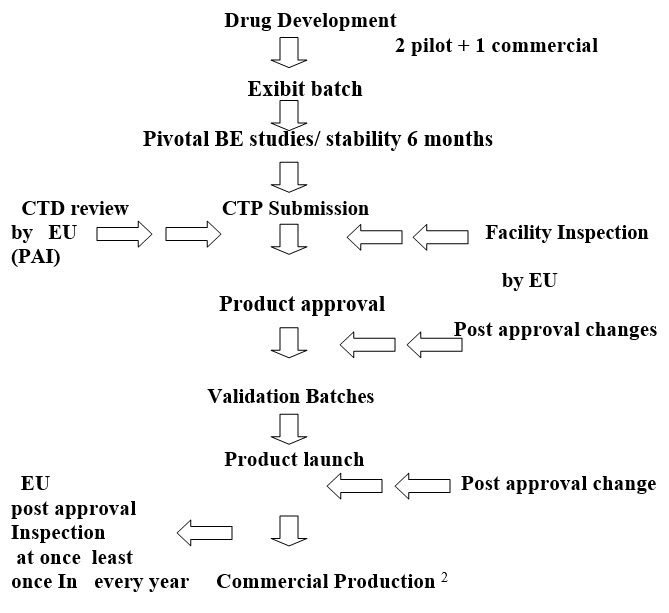

Post approval changes 2
|
Type IA (Minor Changes) |
Deletion of test procedure. If an alternative test procedure is already authorized. |
|
|
Change in imprinting embossing & other marking. |
|
|
Change in the dimension or shape in the immediate release solid dosage form. |
|
|
Change in coating weight of immediate Release product. |
|
|
Minor changes in the Mfg. process not have significant effect on safety, Quality & efficacy. |
|
Type IB (Minor Change) |
Addition or replacement test procedure. |
|
|
Change in the dimension or shape in the prolong release solid dosage form. |
|
|
Replacement of a single excipient with the a comparable one with the same functional Characteristic at same level. |
|
|
Addition or replacement of in process test. |
|
Type II (Major Change) |
Introduction of new manufacture of API change in scoring, break line intended to divide into equal doses. |
|
|
Qualitative or quantitative change in one or more excipient that may have significant impact on safety quality & efficiency of the product deletation of in process test. |
|
|
Change in the coating weight of the prolong release products. |
|
|
Change in the Mfg. process has significant effect on safety, quality & efficacy. |
Administrative2
|
Requirement |
European Union |
|
Application |
MAA |
|
Approval timeline |
12 months |
|
Debarment Certificate |
Not required |
|
Pharmacovigilance |
Required |
|
Agent authorization |
Not required |
Manufacturing and control2
|
Requirement |
European Union |
|
No. of batches |
3 |
|
Process validation |
PV scheme |
|
Executed BMR |
Not required |
|
Comparability Protocol |
Not required |
|
Certificate of suitability |
Mandatory |
|
TSE/ BSE Free statement |
Mandatory |
Finished Product Control2
|
Requirement |
European Union |
|
Assay |
95-105% |
|
Identification test |
Addition test required |
|
Color identification |
Required |
|
Water content |
Not required |
|
Disintegration test |
Required |
Labeling2
|
Requirement |
European Union |
|
NDC no. (National Drug Code) |
Not required |
|
Prescription status |
POM (Prescription only medicine) |
|
Label |
Vials/ cartons |
|
Side by side Comparison |
Not required |
Stability Requirement2
|
Requirement |
European Union |
|
No. of batches |
2 |
|
Date & time of submission |
6 months accelerated & 6 months long term |
|
Container Orientation |
Do not addressed |
Bioequivalence requirement2
|
Requirement |
European Union |
|
CRO |
Audited by MHRA |
|
Reserve sample |
No such requirement |
|
Fasted / Fed |
No such requirement |
|
Retention of sample |
No such requirement but usually follow |
Abbreviation:
POM: Prescription only medicine
NDC: National Drug Code
MAA: Marketing Authorization Application
CTD: Common Technical Documents
CHMP: Committeeon Herbal Medicinal Products
COMP: Committee for Orphan Medicinal Products
CPMP:Committee for Proprietary Medicinal Products
IMPD: Investigational Medicinal Product Dossier
MHRA: Medicines and Healthcare products Regulatory Agency
Result & Discussion:
From the analysis of new drug registration process in European Union study says that it takes approximately 33 to 35 weeks to take approval in European Union. Rules and regulation for clinical trial and marketing authorization are very strict. Each & every company should follow the stages, rules & regulation which are given by government of European Union.
Conclusion:
After studying & analysis the process of new drug registration in European Union its take approximately 33 to 35 weeks. Each & every company should follow the stages, rules & regulation which are given by government of European Union. For approval of new drug first file the application for clinical trial after that apply for marketing authorization of new drug in European Union.
References
1.European Commission Enterprise Directorate-General. (2005, October). Detailed guidance for the request of authorisation of a clinical trial on a medicinal product for human use to the competent authorities, notification of substantial amendments and declaration of the end of the trial. Retrieved March 2, 2010, from docsrush.net/1539908/clinical-trial-authorization-process-eu-uk.html
2. authorstream.com/Presentation/girishswami-1759651-us-eu-submission-comparative/
3. ec.europa.eu/health/authorisation-procedures-centralised_en.htm
4.Clinical Trial Authorizations process of EU.
URL: pharmainfo.net/files/images/stories/article_images/ClinicalTrialAuthorizationProcessofEU.JPG
5. marsdd.com/dms/entrepreneurtoolkit/Regulatory-PDFs/How_New_Drugs_Are_Approved_in_Europe.pdf
REFERENCE ID: PHARMATUTOR-ART-2189
|
PharmaTutor (ISSN: 2347 - 7881) Volume 2, Issue 6 Received On: 14/04/2014; Accepted On: 23/04/2014; Published On: 01/06/2014 How to cite this article: Y Viradiya, M Dagwar, S Lanjewar; Regulatory Affairs: “Study Report of New Drug Registration Process in European Union”; PharmaTutor; 2014; 2(6); 95-107 |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






