

{ DOWNLOAD AS PDF }
ABOUT AUTHORS
VINOTH KUMAR P 1, *RAJALAKSHMI A.N.1, STEPHEN P2
1Department of Pharmaceutics, College Oof Pharmacy, Mother Theresa Postgraduate And Research Institute of Health Sciences, Puducherry-6, India
1 Department of Pharmaceutics, College of Pharmacy, Mother Theresa Postgraduate And Research Institute of Health Sciences, Puducherry-6, India
2 Formulation Research and Development, Sai Mirra Innopharm Pvt Ltd., Chennai, India
*ocusertraji@gmail.com
ABSTRACT:
Orodispersible liquisolid system is the combination of liquisolid technique and orodispersible system. The poor dissolution rate of water insoluble drug is a major drawback for the development of pharmaceutical dosage forms. The oral absorption of drug is most often controlled by dissolution in the gastrointestinal tract. Liquisolid system has been used to enhance dissolution rate of poorly water-soluble drugs. Orodispersible tablets are given in order to provide fast action by disperse in the mouth, without the need of water and make them compliance for paediatric and geriatric patients and to bypass the liver metabolism.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-2591
|
PharmaTutor (Print-ISSN: 2394 - 6679; e-ISSN: 2347 - 7881) Volume 6, Issue 6 Received On: 18/04/2018; Accepted On: 17/05/2018; Published On: 01/06/2018 How to cite this article: Vinoth P, Rajalakshmi AN, Stephen P; Orodispersible liquisolid compacts: A novel approach to enhance solubility and bioavailability; PharmaTutor; 2018; 6(6); 28-35; http://dx.doi.org/10.29161/PT.v6.i6.2018.28 |
INTRODUCTION
The oral route remains the preferred route of drug administration due to its convenience, good patient compliance and low production costs. In order for a drug to be absorbed into the systemic circulation following oral administration, the drug must be dissolved in the gastric fluids (Munke and Nagarsenker, 2004). For hydrophobic drugs, the dissolution process acts as the rate-controlling step and which determines the rate and degree of absorption. Bioavailability of poorly water soluble drugs is limited by their solubility and dissolution rate (Nazzal and Khan, 2006)
Liquisolid technique is a new and promising method that can change the dissolution rate of drugs. It has been used to enhance dissolution rate of poorly water-soluble drugs. The new ‘liquisolid’’ technique may be applied to formulate liquid medications (i.e. oily liquid drugs and solutions, suspensions or emulsions of water-insoluble solid drugs carried in non-volatile liquid vehicles) into powders suitable for tableting or encapsulation. Since, the liquisolid tablets contain a solution of the drug in suitable solvent; the drug surface available for dissolution is tremendously increased (Javadzadeh and Nokhodchi, 2008)
The concept of orodispersible tablet emerged with an objective to improve patient’s compliance. These dosage forms rapidly disintegrate and/or dissolve to release the drug as soon as they come in contact with saliva, thus without the need for water during administration, an attempts that makes them highly attractive for paediatric and geriatric patients (Kaushik et al 2004). When put in the mouth, these dosage forms disintegrate instantly to release the drug, which dissolves or disperses in the saliva. Thereafter, the drug may get absorbed from the pharynx and oesophagus or from other sections of GIT as the saliva travels down. In such cases, bioavailability is significantly greater than that observed from conventional tablet dosage form (Mudgal and Singhai, 2011)
LIQUISOLID SYSTEM
This technique was first introduced by Spireas et al. and applied to incorporate water insoluble drugs into rapid release solid dosage forms. The term “liquisolid compacts” as described by Spireas et.al indicates that immediate or sustained release tablets or capsules that are prepared using the technique of “liquisolid systems” combined with the inclusion of appropriate adjuvants required for tabulating or encapsulation such as lubricants and for rapid or sustained release action, such as disintegrate or binders, respectively (Shashidher and Veer,2012)
CONCEPT OF LIQUISOLID SYSTEM
When the drug dissolved in the liquid vehicle is incorporated into a carrier material which has a porous surface and closely matted fibres in its interior such as celluloses, both absorption and adsorption take place. The liquid initially absorbed in the interior of the particles is captured by its internal structure. After the saturation of this process, adsorption of the liquid onto the internal and external surfaces of the porous carrier particles occurs. Then, the coating material having high adsorptive properties and large specific surface area provides the liquisolid system the desirable flow characteristics (Sambasiva and Naga, 2011).
In liquisolid systems, the drug is already in solution form in liquid vehicle, while at the same time, it is carried by powder. The wettability of the compacts in the dissolution media is one of the proposed mechanisms for explaining the enhanced dissolution rate from the liquisolid compacts.
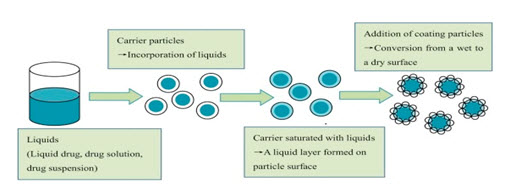
FIG-1:THEORETICAL CONCEPT OF LIQUISOLID SYSTEM
Non-volatile solvent present in the liquisolid system facilitates wetting of drug particles by decreasing interfacial tension between dissolution medium and tablet surface. Thus, due to substantial increase in wettability and effective surface area for dissolution, liquisolid compacts may be expected to reveal enhanced release profiles of water-insoluble drugs.
MATHEMATICAL MODEL TO DESIGN LIQUISOLID SYSTEM:
For getting good flow behaviour and compressibility of liquisolid systems, a mathematical model designed by Spireas et al. was used as formulation design model for the liquisolid tablets (Bhise et al., 2009). Prerequisites for this include suitable drug candidate, suitable non-volatile solvent, carrier and coating materials. The amounts of excipients (carrier and coating materials) used to prepare liquisolid compacts depend on the flowable liquid retention potential values (Ф-value) and the liquid loading factors (Lf).
Flowable liquid retention potential values (Ф- value)
The flowable liquid retention potential (Φ-value) of a powder is defined as the maximum amount of a given non-volatile liquid that can be retained inside its bulk (w/w) while maintaining acceptable flowability.
Therefore, in order to calculate the quantity of excipients, we need to determine the liquid retention potential value for both carrier (ФCA-value) and coating (ФCO-value) materials for each formulation. These values are constant for the given vehicle/powder system.
Liquid loading factors (Lf)
It is defined as the weight ratio of the liquid formulation (W) and the carrier material (Q) in the system:
Lf = W/Q------ (1)
(W is the weight of the liquid medication (the drug + non-volatile liquid vehicle) and Q is the weight of the carrier.)
R represents the ratio between the weights of the carrier (Q) and the coating (q) material present in the formulation. Then optimum weight of the coating material (q) could also be obtained (Equation 2).
R =Q/q------ (2)
The liquid load factor that ensures acceptable flowability (Lf) can be determined by:
Lf = ФCA + ФCO (1/R) ----- (3)
By calculating Lf and W, we can calculate the amount of Q and required for the liquisolid system
TABLE-1: LIQUISOLID FORMULATION PARAMETERS OF VARIOUS POWDER EXCIPIENTS WITH COMMONLY USEDLIQUID VEHICLES (Abdul Hasan Sathali and Deepa, 2013)
|
Non-volatile solvents |
Φ-value for carrier |
Φ-value for coating |
|---|---|---|
|
1.Propylene glycol |
0.16 |
3.31 |
|
2.polyethylene glycol400 |
0.005 |
3.26 |
|
3.Tween 80 |
0.003 |
3.95 |
|
4.Cremophor EL |
0.18 |
0.80 |
|
5.capryol 90 |
0.16 |
0.40 |
COMPONENTS OF LIQUISOLID SYSTEMS
The major formulation components of liquisolid compacts are (Yadav et al., 2010)
1. Carrier Material
These are compression-enhancing, relatively large, preferably porous particles possessing a sufficient absorption property which contributes in liquid absorption.
E.g. various grades of cellulose, starch, lactose, sorbitol, Avicel PH 102 and 200, Eudragit RL and RS, amorphous cellulose etc.
2. Coating Material
These are flow-enhancing, very fine (10 nm to 5,000 nm in diameter), highly adsorptive coating particles (e.g., silica of various grades like Cab-O-Sil M5, Aerosil 200, Syloid 244FP etc.) contributes in covering the wet carrier particles and displaying a dry-looking powder by adsorbing any excess liquid.
3. Non-Volatile Solvents
Inert, high boiling point, preferably water-miscible and not highly viscous organic solvent systems. Various non-volatile solvents used for the formulation of liquisolid systems include Polyethylene glycol 200 and 400, glycerin, polysorbate 80 and propylene glycol, polysorbates, glycerin, N, N- dimethylacetamide, fixed oils, etc.
4. Disintegrant
Superdisintegrants increases the rate of drug release, water solubility and wettability of liquisolid granules. Mostly Superdisintegrants like sodium starch glycolate and crosspovidone and croscarmellose sodium are used.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
MECHANISMS OF ENHANCEMENT OF SOLUBILITY AND BIOAVAILABILITY
Several mechanisms of enhanced drug release have been postulated for liquisolid systems (Saharan, 2009). The three main proposed mechanisms include increased surface area of drug available for release, increased aqueous solubility of the drug due to presence of non-volatile vehicle and improved wettability of the drug particles due to co solvent effect of the vehicle used.
Increased Effective Surface Area
If the drug within the liquisolid system is completely dissolved in the liquid vehicle, it is located in the powder substrate still in a solubilized and molecularly dispersed state. Therefore, the surface area of drug available for release is much greater than that of drug particles within directly compressed tablets. Accordingly, with increasing drug content exceeding the solubility limit and thus, increasing fraction of undissolved drug in the liquid vehicle the release rate decreases. With various drugs it could be shown that the release rates are directly proportional to the fraction of the molecularly dispersed drug (FM) in the liquid formulation. FM is defined by Spireas as the ratio between the drug's solubility (Sd) in the given liquid vehicle and the actual drug concentration (Cd) in this vehicle carried by each system. Therefore,
FM = Sd/ Cd (4)
In addition it is thought that the adsorption and absorption of molecularly dispersed drug onto the surface and interior of the carrier particles impart increased effective surface area available for the mass transfer during the drug dissolution process.
Increased Aqueous Solubility
In addition to the first mechanism of drug release enhancement, it is expected that the solubility of the drug might be increased with liquisolid systems. In fact, the relatively small amount of liquid vehicle in a liquisolid compact is not sufficient to increase the overall solubility of the drug in the aqueous medium. However, in the micro-environment of the solid/liquid interface between an individual primary liquisolid particle and the release medium, it is possible that the amount of liquid vehicle diffusing out of a single liquisolid particle together with the drug molecules might be sufficient to increase the aqueous solubility of the drug if the liquid vehicle can act as a cosolvent.
Improved Wetting Properties
Due to the fact that the liquid vehicle can either act as surface active agent or has a low surface tension, wetting of the primary liquisolid particles is improved.

FIG-2: WETTING PROPERTY OF LIQUISOLID SYSTEM.
Wettability of these systems can be demonstrated by contact angles and water rising times. Also the adsorption of the drug on the carrier particles increases the effective surface area, improving the contact of drug and wettability.
ADVANTAGES
Liquisolid tablets have many advantages. These include:
• Liquisolid systems are low cost formulations than soft gelatine capsules.
• Drug release can be modified using suitable formulation ingredients.
• Drug can be molecularly dispersed in the formulation.
• Capability of industrial production is also possible.
• Enhanced bioavailability can be obtained as compared to conventional tablets.
• Several slightly and very slightly water-soluble and practically water-insoluble liquid and solid drugs can be formulated into liquisolid systems (Dokoumetzidis and Macheras, 2006)
• Even though the drug is in a tablet or capsule form, it is held in a solubilised liquid state, which contributes to increased drug wetting properties, thereby enhancing drug dissolution.
• Rapid release liquisolid tablets or capsules of water insoluble drugs exhibit enhanced In-vitro and in-vivo drug release when compared to their commercial counter parts, including soft gelatin capsules preparation. (Nagabandi et al., 2011)
• Sustained release liquisolid tablets or capsules of water insoluble drugs exhibit constant dissolution rates (zero-order release) comparable only to expensive commercial preparations that combine osmotic pump technology and laser-drilled tablets.
• Better availability of an orally administered water insoluble drug.
• Production of liquisolid systems is similar to that of conventional tablets.
• Can be used for formulation of liquid oily drugs.
• Can be used in controlled drug delivery.
LIMITATIONS
• Not applicable for formulation of high dose insoluble drugs.
• If more amount of carrier is added to produce free-flowing powder, the tablet weight increases to more than one gram which is difficult to swallow.
• Acceptable compression properties may not be achieved since during compression liquid drug may be squeezed out of the liquisolid tablet resulting in tablets of unsatisfactory hardness.
APPLICATIONS
• Liquisolid compact technology is a powerful tool to improve bioavailability of water insoluble drugs. Several water insoluble drugs on dissolving in different non-volatile solvents have been formulated into liquisolid compacts (Gubbi and Jarag, 2009)
• Different drugs can be incorporated into liquisolid compacts.
• Rapid release rates.
• Used for water insoluble solid drugs or liquid lipophilic drugs.
• Sustained release of drugs
• Solubility and dissolution improvement.
• Flowability and compressibility.
• Designing of controlled release tablets.
• Bioavailability enhancement.
• Application in probiotics.
• Improvement of drug photostability.
ORODISPERSIBLE TABLETS
The US Food and Drug Administration Center for Drug Evaluation and Research (CDER) defines, in the ‘Orange Book’, an ODT as “a solid dosage form containing medicinal substances, which disintegrates rapidly, usually within a matter of seconds, when placed upon the tongue”. The significance of these dosage forms is highlighted by the adoption of the term, “Orodispersible Tablet”, by the European Pharmacopoeia which describes it as a tablet that can be placed in oral cavity where it disperses rapidly before swallowing.
DRUG SELECTION CRITERIA FOR ODTs
• Able to saturate the oral mucosa.
• At least moderately non-ionized at oral cavity pH.
• Have the ability to diffuse and partition into the epithelium of upper GIT.
• Small to moderate molecular weight
• Low dose drugs mostly less than 50 mg.
• Should have good stability in saliva and water.
• Drugs with lower bio availability are good candidates for ODT.
• Frequent dosing drugs are unsuitable for ODT.
CHALLENGES AND LIMITATIONS FOR ODTs
• Drugs with relatively larger doses are difficult to formulate into ODTs e.g. antibiotics like ciprofloxacin with adult dose tablet containing about 500 mg of the drug (Velmurugan and Vinushitha, 2010).
• Mechanical strength - ODTs are made of porous or soft molded matrices in order to allow its disintegration in mouth. This makes tablet friable and handling becomes difficult.
• Orodispersible tablets with highly porous structure and good mechanical strength have been developed by sublimation method.
• Palatability - ODTs are intended to be dissolved in mouth. Most of the drugs have bitter taste. Bitter taste can be masked with enough sweetener and flavors.
• Drugs in form of ODTs are hygroscopic in nature and hence need to be protected from humidity (Kumar et al., 2012). To overcome humidity problem special working facilities can be designed by simple methods and special air-conditioning systems can be set up.
• Size of tablet -7mm is easy to swallow while tablets of size 8mm are easy to handle. Hence, tablet sizes which are both easy to handle and swallow are difficult to achieve. For the patient compliance, to make the swallowing easier, round shape punches having optimum dimensions are used.
• Drug candidates should be stable both in water and in saliva, should not ionize at oral cavity pH and should be able to permeate oral mucosal tissue to diffuse and partition in upper GI epithelium (logP > 1, or preferably > 2, should not have short half-life). To optimize solubility problem of the active pharmaceutical ingredient some solid buffers and surfactants can also be chosen.
• ODT requires special packaging for proper stabilization and safety of stable product (Reddy et al., 2012)
ADVANTAGES
1. Administration to the patients who cannot swallow, such as the elderly, stroke victims, bedridden patients, patients affected by renal failure & patients who refuse to swallow such as paediatric, geriatric & psychiatric patients (Shu T, 2002)
2. Rapid drug therapy.
3. Increased bioavailability/rapid absorption through pregastric absorption of drugs from mouth, pharynx & oesophagus as saliva passes down (Chang et al., 2002)
4. Better patient compliance for disabled, bedridden patients, travellers and busy people, who do not always have access to water.
5. Good mouth feel property helps to change the perception of medication as bitter pill particularly in paediatric patients.
6. The risk of chocking during oral administration of conventional formulations due to physical obstruction is avoided, thus providing improved safety.
METHOD OF PREPARATION OF ORODISPERSIBLE LIQUISOLID SYSTEM
As shown in figure-3, a liquid drug can be converted into a dry liquisolid system without being further chemically modified. If liquisolid system of a solid water-insoluble drug is to be formulated, it should be initially dissolved or suspended in a suitable non-volatile solvent system to produce a drug solution or drug suspension of desired concentration (Hamsanandini et al., 2015)
Next, a certain amount of the prepared drug solution or suspension or a liquid drug itself is incorporated into a specific quantity of carrier material which should be preferably of a porous nature and possessing sufficient absorption properties.
The resulting wet mixture is then converted into a dry, non adherent, free-flowing and readily compressible powder by the simple addition and mixing of a calculated amount of coating material
Excipients possessing fine and highly adsorptive particles are suitable for this step. Before compression or encapsulation, various adjuvants like lubricants and super-disintegrants added to final liquisolid system to produce orodispersible liquisolid compacts.
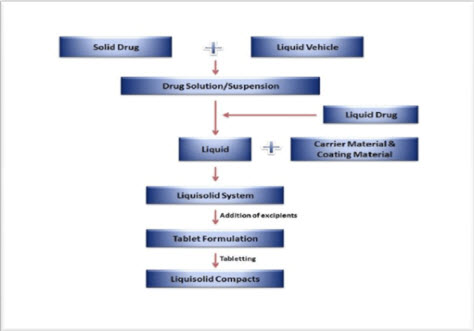
FIG-3: STEPS INVOLVED IN THE PREPARATION OF LIQUISOLID SYSTEM
PRE-FORMULATION STUDIES
1. Solubility studies
Solubility studies are carried out by preparing saturated solutions of drug in non-volatile solvent and analyzing them spectrophotometrically. Saturated solutions are prepared by adding excess of drug to non volatile solvent and shaking them on shaker for specific time period under constant vibration. After this, the solutions are filtered and analyzed spectrophotometrically (Spireas and Sadu, 1998)
2. Determination of angle of slide
Angle of slide is used as a measure of the flow properties of powders. Determination of angle of slide is done by weighing the required amount of carrier material and placed at one end of a metal plate with a polished surface. The end is gradually raised till the plate becomes angular to the horizontal at which powder is about to slide. This angle is known as angle of slide. Angle of 33º is regarded as optimum (Tayel et al., 2008)
3. Determination of flowable liquid retention potential (Φ-value)
The term "flowable liquid- retention potential" (Φ-value) of a powder material describes its ability to retain a specific amount of liquid while maintaining good flow properties (Spireas and Bolton, 1999). The Φ-value is defined as the maximum weight of liquid that can be retained per unit weight of the powder material in order to produce an acceptably flowing liquid/powder admixture. The Φ values are calculated according to equation
Φ value = weight of liquid / weight of solid … (5)
4. Calculation of liquid load factor (Lf)
Different concentrations of non-volatile solvents are taken and the drug is dissolved. Such liquid medication is added to the carrier-coating material admixture and blended (Spireas, 2002). Using equation (6) drug loading factors are determined and used for calculating the amounts of carrier and coating materials in each formulation.
Lf = weight of liquid medication / weight of carrier material… (6)
5. Liquisolid compressibility test (LSC)
The Liquisolid compressibility test is used to determine Φ values and involves steps such as preparing carrier coating material admixture systems, preparing several uniform liquid or powder admixtures, compressing each liquid or powder admixtures to tablets, assessing the average hardness, determination of the average liquid content of crushed tablets, as well as determining plasticity, sponge index and Φ value and Lf.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
SOLUBILITY AND DISSOLUTION IMPROVEMENT:
This technique was successfully applied for low dose water insoluble drugs. When the therapeutic dose of drug is more than 50mg, dissolution enhancement in the presence of low levels of hydrophilic carrier and coating material is not significant. But by adding some materials such as polyvinyl pyrrolidone (PVP) to liquid medication (microsystems), it would be possible to produce dry powder formulations containing liquid with high concentration of drug. By adding such materials to the liquid medication, low amount of carrier is required to obtain dry powder with free flowability and good compatibility (Amrit et al., 2009).
BIOAVAILABILITY IMPROVEMENT:
In the liquisolid and powdered solution systems the drug might be in a solid dosage form, it is held within the powder substrate in solution, or in a solubilized, almost molecularly dispersed state. Therefore, due to their significantly increased wetting properties and surface of drug available for dissolution, liquisolid compacts of water insoluble substances may be expected to display enhanced drug release properties, and consequently, Improved bioavailability (Yadav VB and Yadav AV., 2009).
CONCLUSION
The Liquisolid system is a technique for formulation of water insoluble drugs to enhance their aqueous solubility, absorption as well as dissolution rate which leading to enhancement of bioavailability of drugs as compared to conventional directly compressed tablets. Orodispersible tablets may give rapid onset of action by rapid absorption through pre-gastric absorption of drug from mouth, pharynx and oesophagus as saliva passes down and beneficial to reduce dose. By combining Liquisolid technique and Orodispersible DDS, may enhance solubility, dissolution rate by means of Liquisolid technique and can achieve rapid onset of action with lower dose of drug by using Orodispersible DDS and hence may increase patient compliance.
REFERENCES:
1. Abdul Hasan Sathali A., Deepa C (2013); Formulation of liquisolid tablets of candesartan cilexetil; Int. J. Res. Pharm. Sci; 2013; 4(2); 238-249.
2. Amrit B., Karmarkar Indrajeet D.,Gonjari., Avinash H Hosmani., Pandurang N Dhabale., Satish B Bhise (2009); Liquisolid Tablets: A Novel Approach for Drug Delivery; International Journal of Health Research; 2009; 2(1); 45-50.
3. Bhise SB., Nighute AB., Yadav AV., Yadav VB (2009); Aceclofenac size enlargement by non aqueous granulation with improved solubility and dissolution; Arch Pharm Sci & Res; 2009; (1); 115-122.
4 .Chang RK., Guo X., Burnside BA., Cough RA (2002); Fast dissolving tablets; Pharm Tech; 2002; (24); 52-58.
5. Dokoumetzidis A., & Macheras P (2006); A century of dissolution research: from Noyes and Whitney to the biopharmaceutics classification system; International journal of pharmaceutics; 2006; (321); 1 –11.
6. Gubbi S.R., Jarag R (2009); Research J. Pharm. And Tech; 2009; 2(2); 382-386.
7. Hamsanandini J., Parthiban S., Vikneswari A., Sentil kumar G.P., Tamiz Mani T (2015); Formulation and Evaluation of Orodispersible Liquisolid Compacts of Meloxicam using Banana Powder as a Natural Superdisintegrants; Asian Journal of Research in Biological and Pharmaceutical Sciences; 2015; 3(1); 25-38.
8. Javadzadeh Y., Nokhodchi A (2008); Liquisolid technique for dissolution rate enhancement of a high dose water-insoluble drug (carbamazepine); Int J Pharm; 2008; 341;26-34.
9. Kaushik D., Dureja H., Saini TR (2004); Mouth dissolving tablets: A Review; Indian Drugs; 2004; 41(4): 187-193.
10. Kumar S., Gupta SK., Sharma PK (2012); A Review on Recent Trends in Oral Drug Delivery-Fast Dissolving Formulation Technology; Adv Biol Res; 2012; (6); 6-13.
11. Mudgal V.K., singhai A.K (2011); Orally Disintegrating tablet: A Review; International Research Journal of pharmacy; 2011; ISSN 2230-8407.
12. Munke AP., Nagarsenker MS (2004); Triamterene β-cyclodextrin complexes: Preparation, characterization and In vivo evaluation; AAPS PharmSciTech; 2004; (5); 1-9.
13. Nazzal S., Khan MA (2006); Controlled release of a self-emulsifying formulation from a tablet dosage form: Stability assessment and optimization of some processing parameters; Int. J. Pharm; 2006; (315); 110–121.
14. Reddy M., Babu S., Harshita B., Sravya R (2012); Conventional and Patented Technologies in Oral Dispersible Tablets: A Review; J Chem Pharm Sci; 2012; (6); 286-92.
15. Saharan V (2009); Dissolution enhancement of drugs. Part I: technologies and effect of carriers; Int. J. Health Res; 2009; (2); 107-24.
16. Sambasiva AR., Naga TA (2011); Liquisolid Technology: An Overview; Int J Res Pharma Biomed Sci; 2011; 2(2); 401-409.
17. Shashidher B., Veera R (2012); Formulation and evaluation of carvedilol liquisolid tablets; A. j. p. s. P; 2012; 30-44.
18. Shu T (2002); Studies of rapidly disintegrating tablets in oral cavity using co-ground mixture of mannitol with crospovidone; Chem Pharm Bull; 2002; (50); 193-198.
19. Spireas S (2002); Liquisolid Systems and Methods of Preparing Same; U.S. Patent 6423339 B1; 2002.
20. Spireas S., Bolton M (1999); Liquisolid Systems and Methods of Preparing Same; U.S. Patent 5968.1999; 550.
21. Spireas S., Sadu S (1998); Enhancement of prednisolone dissolution properties using liquisolid compacts; Int. J.Pharm; 1998; (166):177–188.
22. Tayel SA., Soliman II., Louis D (2008); Improvement of Dissolution Properties of Carbamazepine through Application of the Liquisolid Technique; Eur J Pharm Biopharm; 2008; (69); 342-347
23. Velmurugan S., Vinushitha S (2010); Oral Disintegrating Tablets: An Overview; Int J Chem Pharm Sci; 2010; 1-12.
24. Vijaykumar Nagabandi., Ramarao T., Jayaveera K.N (2011); Liquisolid Compacts: A Novel Approach to Enhance Bioavailability of Poorly Soluble Drugs; International Journal of Pharmacy and Biological Sciences; 2011; 89-102.
25. Yadav A.V., Shete A.S., Dabke A.P (2010); Indian J. Pharm. Educ. Res; 2010; 44(3); 227-235
26. Yadav VB., Yadav AV (2009); Liquisolid granulation technique for tablet manufacturing: an overview; Journal of Pharmacy Research; 2009; 2(4); 670-674.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






