

 About Authors:
About Authors:
Nilesh Sovasia*, Arshad Hala
Seth G.L.Bihani S.D.College Of Technical Education,
Institute Of Pharmaceutical Science & Drug Research,
Sri Ganganagar, Rajasthan, India
*nilesh.sovasia@yahoo.com
ABSTRACT
The microbiological assay of an antibiotic is based upon a comparison of the inhibition of growth of micro-organisms by measured concentrations of the antibiotics under examination with that produced by known concentrations of a standard preparation of the antibiotic having a known activity.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1560
INTRODUCTION:
Two general methods are usually employed, the cylinder-plate (or cup-plate) method and the turbidimetric (or tube assay) method.
The cylinder-plate method (Method A) depends upon diffusion of the antibiotic from a vertical cylinder through a solidified agar layer in a Petri dish or plate to an extent such that growth of the added micro-organism is prevented entirely in a zone around the cylinder containing a solution of the antibiotic. The turbidimetric method (Method B) depends upon the inhibition of growth of a microbial culture in a uniform solution of the antibiotic in a fluid medium that is favourable to its rapid growth in the absence of the antibiotic.
The assay is designed in such a way that the mathematical model on which the potency equation is based can be proved to be valid. If a parallel-line model is chosen, the two log doseresponse lines of the preparation under examination and the standard preparation should be parallel; they should be rectilinear over the range of doses used in the calculation. These conditions should be verified by validity tests for a given probability. Other mathematical models, such as the slope ratio method, may be used provided that proof of validity is demonstrated.
MEDIA:
Prepare the media required for the preparation of test organism inocula from the ingredients listed in Table 1. Minor modifications of the individual ingredients may be made, or reconstituted dehydrated media may be used provided the resulting media have equal or better growth-promoting properties and give a similar standard curve response.
Dissolve the ingredients in sufficient waterto produce 1000 ml and add sufficient 1 M sodium hydroxideor 1 M hydrochloric acid, as required so that after sterilization the pH is as given in Table 1.
Table 1– Media: Quantities in g of ingredients per 1000 ml
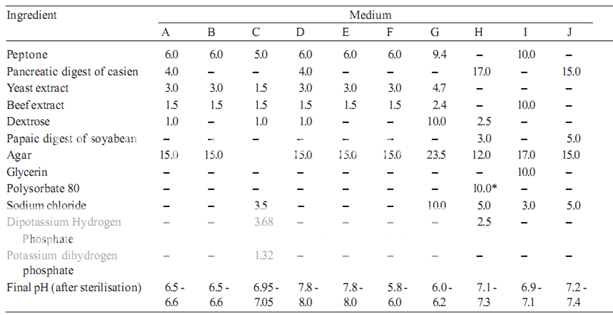
* Quantity in ml, to be added after boiling the media to dissolve the agar.
STANDARD PREPARATION AND UNITS OF ACTIVITY
A Standard Preparation is an authentic sample of the appropriate antibiotic for which the potency has been precisely determined by reference to the appropriate international standard. The Potency of the standard preparation may be expressed in International Units or in μg per mg of the pure antibiotic.
The Standard Preparations for India are certified by the laboratory of the Indian Pharmacopoeia Commission or by any other notified laboratory(ies) and are maintained and distributed by the agency(ies) notified for the purpose.
A Standard Preparation may be replaced by a working standard prepared by any laboratory which should be compared at definite intervals under varying conditions with the standard.
Buffer solution
Prepare by dissolving the following quantities given in Table 2 of dipotassiumhydrogen phosphate and potassium dihydrogen phosphatein sufficient waterto produce 1000 ml after sterilisation, adjusting the pH with 8 M phosphoric acidor 10 M potassium hydroxide.
Table 2 – Buffer Solutions
|
Buffer No. |
Dipotassium Hydrogen Phosphate, K2HPO4(g) |
Potassium Dihydrogen phosphate, KH2PO4(g) |
pH adjusted after sterilization to |
|
1 2 3 4 5 6 |
2.0 16.73 - 20.0 35.0 13.6 |
8.0 0.523 13.61 80.00 - 4.0 |
6.0±0.1 8.0±0.1 4.5±0.1 6.0±0.1 10.5±0.1* 7.0±0.2 |
* After addition of 2 ml of 10M potassium hydroxide
Preparation of the standard solution
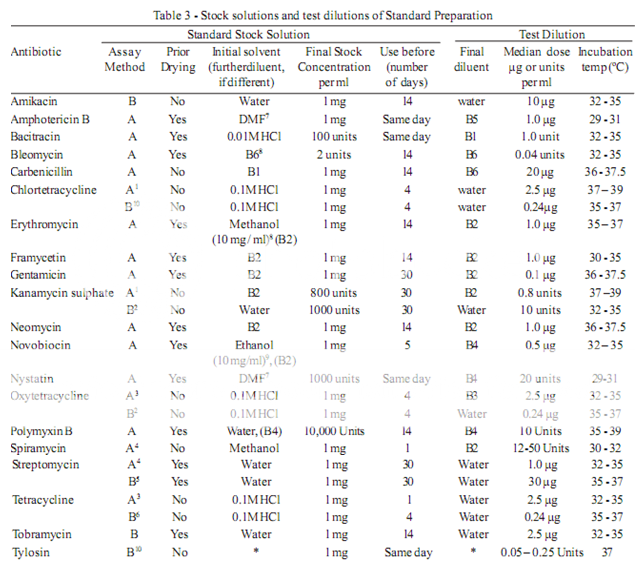
To prepare a stock solution, dissolve a quantity of the Standard Preparation of a given antibiotic, accurately weighed and previously dried where so indicated in Table 3, in the solvent specified in the table, and then dilute to the required concentration as indicated. Store in a refrigerator and use within the period indicated. On the day of assay, prepare from the stock solution five or more test dilutions, the successive solutions increasing stepwise in concentration, usually in the ratio 1:1.25 for Method A or smaller for Method B. Use the final diluent specified and a sequence such that the middle or median has the concentration specified in Table 3
Preparation of the sample solution
From the information available for the substance under examination (the “unknown”), assign to it an assumed potency per unit weight or volume, and on this assumption prepare on the day of the assay a stock solution and test dilution as specified for each antibiotic in Table 3 but with the same final diluent as used for the Standard Preparation. The assay with 5 levels of the Standard requires only one level of the unknown at a concentration assumed equal to the median level of the standard.
Test organisms
The test organism for each antibiotic is listed in Table 4, together with its identification number in the American Type Culture Collection (ATCC). Maintain a culture on slants of the medium and under the incubation conditions specified in Table 5, and transfer weekly to fresh slants.
Table 4 - Test Organisms for Microbiological Assay of Antibiotics
|
Antibiotic |
Test Organism |
ATCC1 No. |
|
Amikacin Amphotericin B Bacitracin Bleomycin Carbenicillin Chlortetracycline Erythromycin Framycetin
Gentamicin Kanamycin sulphate
Neomycin Novobiocin Nystatin Oxytetracycline
Polymyxin B Spiramycin Streptomycin
Tetracycline
Tobramycin Tylosin |
Staphylococcus aureus Saccharomyces cerevisiae Micrococcus luteus Mycobacterium smegmatis Pseudomonas aeruginosa Bacillus pumilus Micrococcus luteus Bacillus pumilus Bacillus subtilis Staphylococcus epidermidis Bacillus pumilus Staphylococcus aureus Staphylococcus epidermidis Staphylococcus epidermidis Saccharomyces cerevisiae Bacillus cereus var, mycoides Staphylococcus aureus Bordetella bronchiseptica Bacillus pumilus Bacillus subtilis Klebsiella pnumoniae Bacillus cereus Staphylococcus aureus Staphylococcus aureus Staphylococcus aureus |
29737 9763 10240 607 25619 14884 9341 14884 6633 12228 14884 29737 12228 12228 2601 11778 29737 4617 6633 6633 10031 11778 29737 29737 9144 |
1. American Type Culture Collection, 21301 Park Lawn Drive, Rockville, MD20852, USA
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
CULTURE MEDIA
Media for the tests may be prepared as described below, or equivalent commercially available dehydrated mixtures yielding similar formulations may be used provided that when reconstituted as directed by the manufacturer, they comply with the growth promotion test. Other media may be used provided that they have been shown to sustain the growth of a wide range of micro-organisms.
The following culture media have been found to be suitable for the test. Fluid thioglycollate medium is primarily intended for the culture of anaerobic bacteria; however, it will also detect aerobic bacteria. Soyabean-casein digest medium is suitable for the culture of both fungi and aerobic bacteria.
Fluid Thioglycollate Medium – For use with clear fluid products.
L-Cystine 0.5 g
Sodium chloride 2.5 g
Dextrose monohydrate/anhydrous 5.5 g/5.0 g
Granular agar (moisture content
less than 15 per cent, w/w) 0.75 g
Yeast extract (water-soluble) 5.0 g
Pancreatic digest of casein 15.0 g
Sodium thioglycollate or 0.5 g
Thioglycollic acid 0.3 ml
Resazurin sodium solution
(0.1 per cent), freshly prepared 1.0ml
Distilled water to 1000 ml
pH of the medium after sterilization 7.1 ± 0.2
Mix the ingredients other than the thioglycollate or thioglycollic acid and the resazurin sodium solution, in the order given above, in a mortar, with thorough grinding. Stir in some heated distilled water, transfer to a suitable container, add the remainder of the distilled water, and complete the solution by heating in a boiling water-bath. Dissolve the sodium thioglycollate or thioglycollic acid in the solution and, if necessary, add 1M sodium hydroxide so that, after sterilisation, the solution will have a pH of 7.1 ± 0.2. If filtration is necessary, heat the solution again without boiling and filter while hot through moistened filter paper. Add the resazurin sodium solution, mix and distribute the medium into suitable vessels that provide a ratio of surface to depth of medium such that not more than the upper half of the medium has undergone a colour change indicative of oxygen uptake at the end of the incubation period. Sterilise in an autoclave at 121º for 20 minutes. If the medium is to be stored, cool promptly to 25º and store at 2º to 30º, avoiding excess of light. If more than the upper one-third of the medium has acquired a pink colour, the medium may be restored once by reheating in a water-bath or in free-flowing steam until the pink colour disappears, and cooling rapidly, taking care to prevent the introduction of non-sterile air into the container. When ready for use, not more than the upper one-tenth of the medium should have a pink colour. Medium more than 4 weeks old should not be used.
Use fluid thioglycollate medium by incubating it at 30º to 35º.
Alternative Thioglycollate Medium — For use with turbid and viscid products and for devices having tubes with small lumina.
L-Cystine 0.5 g
Sodium chloride 2.5 g
Dextrose monohydrate/anhydrous 5.5 g/5.0 g
Yeast extract (water-soluble) 5.0 g
Pancreatic digest of casein 15.0 g
Sodium thioglycollate or 0.5 g
Thioglycollic acid 0.3 ml
Distilled water to 1000 ml
pH of the medium after sterilization 7.1 ± 0.2
Heat the ingredients in a suitable container until solution is effected. Mix, add 1M sodium hydroxide, if necessary, so that, after sterilisation, the medium will have a pH of 7.1 ± 0.2. Filter, if necessary, place in suitable vessels and sterilise at 121º for 20 minutes. Store at a temperature between 2º and 25º in a sterile sealed container, unless it is intended for immediate use.
The medium is freshly prepared or heated in a water-bath and allowed to cool just prior to use. It should not be reheated.
Use alternative thioglycollate medium in a manner that will assure anaerobic conditions for the duration of the incubation at 30º to 35º.
Soyabean-casein Digest Medium
Pancreatic digest of casein 17.0 g
Papaic digest of soyabean meal 3.0 g
Sodium chloride 5.0 g
Dipotassium hydrogen phosphate 2.5 g
(K2HPO4)
Dextrose monohydrate/anhydrous 2.5 g/2.3 g
Distilled water to 1000 ml
pH of the medium after sterilisation 7.3 ± 0.2
Dissolve the solids in distilled water, warming slightly to effect solution. Cool to room temperature and add, if necessary, sufficient 1M sodium hydroxide so that after sterilisation the medium will have a pH of 7.3 ± 0.2. Filter, if necessary, distribute into suitable containers and sterilise in an autoclave at 121º for 20 minutes.
Use soyabean-casein digest medium by incubating it at 20º to 25º under aerobic conditions.
PREPARATION OF THE INOCULUM
Prepare the microbial suspensions for the inoculum for the assay as given in Table 5. If the suspensions are prepared by these methods, growth characteristics are sufficiently uniform so that the inoculum can be adequately determined by the trials given below.

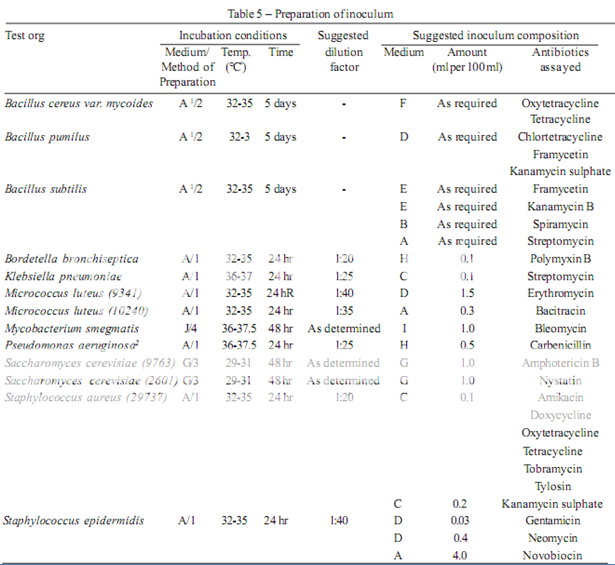
1. Use Medium A containing 300 mg of manganese sulphate per litre.
2. For Pseudomonas aeruginosa in the assay of Carbenicillin, use the dilution yielding 25 per cent light transmission, rather than the stock suspension, for preparing the inoculum suspension.
METHODS OF PREPARATION OF TEST ORGANISM SUSPENSION:
1. Maintain the test organism on slants of Medium A and transfer to a fresh slant once a week. Incubate the slants at the temperature indicated above for 24 hours. Using 3 ml of saline solution, wash the organism from the agar slant onto a large agar surface of Medium A such as a Roux bottle containing 250 ml of agar. Incubate for 24 hours at the appropriate temperature. Wash the growth from the nutrient surface using 50 ml of saline solution. Store the test organism under refrigeration. Determine the dilution factor which will give 25 per cent light transmission at about 530 nm. Determine the amount of suspensions to be added to each 100 ml of agar of nutrient broth by use of test plates or test broth. Store the suspension uder refrigeration.
2. Proceed as described in Method 1 but incubate the Roux bottle for 5 days. Centrifuge and decant the supernatant liquid. Resuspend the sediment with 50 to 70 ml of saline solution and heat the suspension for 30 minutes at 70º. Wash the spore suspension three times with 50 to 70 ml of saline solution. Resuspend in 50 to 70 ml of saline solution and heat- shock again for 30 minutes. Use test plates to determine the amount of the suspension required for 100 ml of agar. Store the suspension under refrigeration.
3. Maintain the test organism on 10 ml agar slants of Medium G. Incubate at 32º to 35º for 24 hours. Inoculate 100 ml of nutrient broth. Incubate or 16 to 18 hours at 37º and proceed as described in Method I.
4. Proceed as described in Method 1 but wash the growth from the nutrient surface using 50 ml of Medium 1 (prepared without agar) in place of saline solution.
DETERMINATION OF INOCULUM
For Method A. After the suspension is prepared as given under Table 5, add different volumes of it to each of several different flasks containing 100 ml of the medium specified in Table 3 (the volume of suspension suggested in Table 3 may be used as a guide). Using these inocula, prepare inoculated plates as described for the specific antibiotic assay. While conducting cylinder-plate assays, double-layer plates may be prepared by pouring a seed layer (inoculated with the desired micro organism) over a solidified uninoculated base layer. For each Petri dish, 21 ml of base layer and 4 ml of the seed layer may be generally suitable. Fill each cylinder with the median concentration of the antibiotic (Table 3) and then incubate the plates. After incubation, examine and measure the zones of inhibition. The volume of suspension that produces the optimum zones of inhibition with respect to both clarity and diameter determines the inoculum to be used for the assay.
For Method B. Proceed as described for Method A and, using the several inocula, carry out the procedure as described for the specific antibiotic assay running only the high and low concentrations of the standard response curve. After incubation, read the absorbances of the appropriate tubes. Determine which inoculum produces the best response between the low and high antibiotic concentrations and use this inoculum for the assay.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
APPARATUS
All equipment is to be thoroughly cleaned before and after each use. Glassware for holding and transferring test organisms is sterilised by dry heat or by steam.
TEMPERATURE CONTROL.
Thermostatic control is required at several stages of a microbial assay, when culturing a microorganism and preparing its inoculum and during incubation in a plate assay. Closer control of the temperature is imperative during incubation in a tube assay which may be achieved by either circulated air or water, the greater heat capacity of water lending it some advantage over circulating air.
SPECTROPHOTOMETER.
Measuring transmittance within a fairly narrow frequency band requires a suitable spectrophotometer in which the wavelength of the light source can be varied or restricted by the use of a 580-nm filter for preparing inocula of the required density or with a 530-nm filter for reading a absorbance in a tube assay. For the latter purpose, the instrument may be arranged to accept the tube in which incubation takes place, to accept a modified cell fitted with a drain that facilitates rapid change of contents, or preferably fixed with a flow-through cell for a continuous flow-through analysis. Set the instrument at zero absorbance with clear, uninoculated broth prepared as specified for the particular antibiotic, including the same amount of test solution and formaldehyde as found in each sample.
Cylinder-plate assay receptacles. Use rectangular glass trays or glass or plastic Petri dishes (approximately 20 x 100 mm) having covers of suitable material and assay cylinders made of glass, porcelain, aluminium or stainless steel with outside diameter 8 mm ± 0.1 mm, inside diameter 6mm ± 0.1mm and length 10 mm ± 0.1 mm. Instead of cylinders, holes 5 to 8 mm in diameter may be bored in the medium with a sterile borer, or paper discs of suitable quality paper may be used. Carefully clean the cylinder to remove all residues. An occasional acidbath, e.g. with about 2 M nitric acid or with chromic acid solution is needed.
Turbidimetric assay receptacles.
For assay tubes, use glass or plastic test-tubes, e.g. 16 mm x 125 mm or 18 mm x 150 mm that are relatively uniform in length, diameter, and thickness
and substantially free from surface blemishes and scratches. Cleanse thoroughly to remove all antibiotic residues and traces of cleaning solution and sterilise tubes that have been used previously before subsequent use.
ASSAY DESIGNS
Microbial assays gain markedly in precision by the segregation of relatively large sources of potential error and bias through suitable experimental designs. In a cylinder plate assay, the essential comparisons are restricted to relationships between zone diameter measurements within plates, exclusive of the variation between plates in their preparation and subsequent handling. To conduct a turbidimetric assay so that the difference in observed turbidity will reflect the differences in the antibiotic concentration requires both greater uniformity in the environment created for the tubes through closer thermostatic control of the incubator and the avoidance of systematic bias by a random placement of replicate tubes in separate tube racks, each rack containing one complete set of treatments. The essential comparisons are then restricted to relationships between the observed turbidities within racks. Within these restrictions, two alternative designs are recommended; i.e. a 3-level (or 2-level) factorial assay, or a 1- level assay with a standard curve. For a factorial assay, prepare solutions of 3 or 2 corresponding test dilutions for both the standard and the unknowns on the day of the assay, as described under Preparation of the Standard and Preparation of the samples. For a 1-level assay with a standard curve, prepare instead solutions of five test dilutions of the standard and a solution of a single median test level of the unknown as described in the same sections. Consider an assay as preliminary if its computed potency with either design is less than 60 per cent or more than 150 per cent of that assumed in preparing the stock solution of the unknown. In such a case, adjust its assumed potency accordingly and repeat the assay. Microbial determinations of potency are subject to inter-assay variables as well as intra-assay variables, so that two or more independent assays are required for a reliable estimate of the potency of a given assay preparation or unknown. Starting with separately prepared stock solutions and test dilutions of both the standard and unknown, repeat the assay of a given unknown on a different day. If the estimated potency of the second assay differs significantly, as indicated by the calculated standard error, from that of the first, conduct one or more additional assays. The combined result of a series of smaller, independent assays spread over a number of days is a more reliable estimate of potency than that from a single large assay with the same total number of plates or tubes.
METHODS
Carry out the microbiological assay by Method A or Method B.
A. Cylinder-plate or Cup-plate method
Inoculate a previously liquefied medium appropriate to the assay (Tables 1 and 3) with the requisite quantity of suspension of the micro organism, add the suspension to the medium at a temperature between 40º and 50º and immediately pour the inoculated medium into the petri dishes or large rectangular plates to give a depth of 3 to 4 mm (1 to 2mm for nystatin). Ensure that the layers of medium are uniform in thickness, by placing the dishes or plates on a level surface.
Store the prepared dishes or plates in a manner so as to ensure that no significant growth or death of the test organism occurs before the dishes or plates are used and that the surface of the agar layer is dry at the time of use.
Using the appropriate buffer solutions indicated in Tables 2 and 3, prepare solutions of known concentrations of the standard preparation and solutions of the corresponding assumed of concentrations the antibiotic to be examined. Where directions have been given in the individual monograph for preparing the solutions, these should be followed and further dilutions made with buffer solution as indicated in Table 3. Apply the solutions to the surface of the solid medium in sterile cylinders or in cavities prepared in the agar. The volume of solution added to each cylinder or cavity must be uniform and sufficient almost to fill the holes when these are used. When paper discs are used these should be sterilized by exposure of both sides under a sterilising lamp and then impregnated with the standard solutions or the test solutions and placed on the surface of the medium. When Petri dishes are used, arrange the solutions of the Standard Preparation and the antibiotic under examination on each dish so that, they alternate around the dish and so that the highest concentrations of standard and test preparations are not adjacent. When plates are used, place the solutions in a Latin square design, if the plate is a square, or if it is not, in a randomised block design. The same random design should not be used repeatedly.
Leave the dishes or plates standing for 1 to 4 hours at room temperature or at 4º, as appropriate, as a period of preincubation diffusion to minimise the effects of variation in time between the application of the different solutions. Incubate them for about 18 hours at the temperature indicated in Table 3. Accurately measure the diameters or areas of the circular inhibition zones and calculate the results.
Selection of the assay design should be based on the requirements stated in the individual monograph. Some of the usual assay designs are as follows.
(a) One-level assay with standard curve
Standard Solution. Dissolve an accurately weighed quantity of the Standard Preparation of the antibiotic, previously dried where necessary, in the solvent specified in Table 3, and then dilute to the required concentration, as indicated, to give the stock solution. Store in a refrigerator and use within the period indicated. On the day of the assay prepare from the stock solution, 5 dilution (solutions S1 to S5) representing 5 test levels of the standard and increasing stepwise in the ratio of 4:5. Use the diluent specified in Table 3 and a sequence such that the middle or median has the concentration given in the table.
Sample Solution. From the information available for the antibiotic preparation which is being examined (the “unknown”) assign to it an assumed potency per unit weight or volume and on this assumption prepare on the day of the assay a stock solution with same solvent as used for the standard. Prepare from this stock solution a dilution to a concentration equal to the median level of the standard to give the sample solution.
Method. For preparing the standard curve, use a total of 12 Petri dishes or plates to accommodate 72 cylinders or cavities. A set of 3 plates (18 cylinders or cavities) is used for each dilution. On each of the three plates of a set fill alternate cylinders or cavities with solution S3 (representing the median concentration of the standard solution) and each of the remaining 9 cylinders or cavities with one of the other 4 dilutions of the standard solution. Repeat the process for the other 3 dilutions of the standard solution. For each unknown preparation use a set of 3 plates (18 cylinders or cavities) and fill alternate cylinders or cavities with the sample solution and each of the remaining 9 cylinders of cavities with solution S3. Incubate the plates for about 18 hours at the specified temperature and measure the diameters or the zones of inhibition.
Estimation of potency.
Average the readings of solution S3 and the readings of the concentration tested on each sets of three plates, and average also all 36 readings of solution S3. The average of the 36 readings of solution S3 is the correction point for the curve. Correct the average value obtained for each concentration (S1, S2, S4 and S5) to the figure it would be if the readings for solution S3 for that set of three plates were the same as the correction point. Thus, in correcting the value obtained with any concentration, say S1, if the average of 36 readings of S3 is, for example, 18.0 mm and the average of the S3 concentrations on one set of three plates is 17.8 mm, the correction is + 0.2 mm. If the average reading of S1 is 16.0 mm the corrected reading of S1 is 16.2 mm. Plot these corrected values including the average of the 36 readings for solutions S3 on two-cycle semilog paper, using the concentrations in Units or μg per ml (as the ordinate logarithmic scale) and the diameter of the zones of inhibition as the abscissa. Draw the straight response line either through these points by inspection or through the points plotted for highest and lowest zone diameters obtained by means of the following expressions:
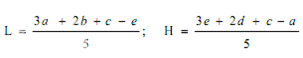
where, L = the calculated zone diameter for the lowest concentration of the standard curve response line.
H = the calculated zone diameter for the highest concentration of the standard curve response line.
c = average zone diameter of 36 readings of the reference point standard solution.
a,b,d,e = corrected average values for the other standard solutions, lowest to highest concentrations, respectively.
Average the zone diameters for the sample solution and for solutions S3 on the plates used for the sample solution. If sample gives a large average zone size than the average of the standard (solution S3), add the difference between them to the zone size of solution S3 of the standard response line. If the average sample zone size is smaller than the standard values, subtract the difference between them from the zone size of solution S3 of the standard response line. From the response line read the concentration corresponding to these corrected values of zone sizes. From the dilution factors the potency of the sample may be calculated.
(b) Two-level factorial assay
Prepare parallel dilutions containing 2 levels of both the standard (S1 and S2) and the unknown (U1and U2). On each of four or more plates, fill each of its four cylinders or cavities with a different test dilution, alternating standard and unknown. Keep the plates at room temperature and measure the diameters of the zones of inhibition.
Estimation of potency. Sum the diameters of the zones of each dilution and calculate the percentage potency of the sample (in terms of the standard) from the following equation :
Per cent potency = Antilog (2.0 + a log I
where in a may have a positive or negative value and should be used algebraically and
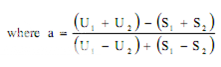
U1 and U2 are the sums of the zone diameters with solutions of the unknown of high and low levels.
S1 and S2 are the sums of the zone diameters with solutions of the standard of high and low levels.
I = ratio of dilutions.
If the potency of the sample is lower than 60 per cent or greater that 150 per cent of the standard, the assay is invalid and should be repeated using higher or lower dilutions of the same solution. The potency of the sample may be calculated from the expression.
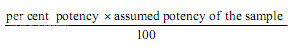
(c) Other designs
1. Factorial assay containing parallel dilution of three test levels of standard and the unknown.
2. Factorial assay using two test levels of standard and two test levels of two different unknowns.
B. Turbidimetric or Tube assay method
The method has the advantage of a shorter incubation period for the growth of the test organism (usually 3 to 4 hours) but the presence of solvent residues or other inhibitory substances affects this assay more than the cylinder plates assay and care should be taken to ensure freedom from such substances in the final test solutions. This method is not recommended for cloudy or turbid preparations.
Prepare five different concentrations of the standard solution for preparing the standard curve by diluting the stock solution of the Standard Preparation of the antibiotic (Table 3) and increasing stepwise in the ration 4:5. Select the median concentration (Table 3) and dilute the solution of the substance being examined (unknown) to obtain approximately this concentration. Place 1 ml of each concentration of the standard solution and of the sample solution in each of the tubes in duplicate. To each tube add 9 ml of nutrient medium (Table 3) previously seeded with the appropriate test organism (Table 3).
At the same time prepare three control tubes, one containing the inoculated culture medium (culture control), another identical with it but treated immediately with 0.5 ml of diluteformaldehyde solution (blank) and a third containing uninoculated culture medium.
Place all the tubes, randomly distributed or in a randomized block arrangement, in an incubator or water-bath and maintain them at the specified temperature (Table 3) for 3 to 4 hours. After incubation add 0.5 ml of dilute formaldehyde solution to each tube. Measure the growth of the test organism by determining the absorbance at about 530 nm of each of the solutions in the tubes against the blank (2.4.7).
Estimation of potency. Plot the average absorbances for each absorbances on the arithmetic scale and concentrations on the logarithmic scale. Construct the best straight response line through the points either by inspection or by means of the following expressions:
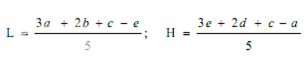
where, L= the calculated absorbance for the lowest concentration of the standard response line.
H= the calculated absorbance for the highest concentration of the standard response line.
a, b, c, d, e = average absorbance values for each concentration of the standard response line lowest to highest respectively.
Plot the values obtained for L and H and connect the points. Average the absorbances for the sample and read the antibiotic concentration from the standard response line. Multiply the concentration by the appropriate dilution factors to obtain the antibiotic content of the sample.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
EXAMPLE OF ANTIMICROBIAL ASSAY OF TETRACYCLINE IN MILK
Introduction
By definition, an antibiotic is either a natural product of a micro-organism, an identical synthetic product or a similar semi synthetic product, that inhibits the growth of other microorganisms (bacteriostatic effect) or destroys other microorganisms (bactericide effect).
In the field of food hygiene and food control we deal with analysis of antibiotic residues in food of animal origin due to the potential of unwanted consequences . Among them are sensitivity to antibiotics, allergic reactions and imbalance of intestinal microflora in people, spread of resistance to antibiotics in microorganisms and losses in the food industry where antibiotics can influence starter cultures used in the production of meat and milk products.
Microbial methods were the first choice of systematic detection of antibiotic residues in food in the past and are still mainstream screening methods. They allow determination of the presence of antibiotics in the sample and identification of specific antibiotic groups.
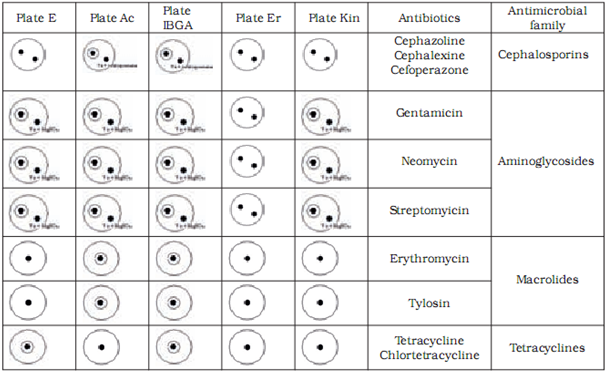
The most frequently used microbial method is based on the principle of inhibition of growth of testing strains which is known as the STAR five-plate method.It is used for detection of antibiotics from the macrolide, aminoglicoside, tetracycline and cephalosporine families.
Material and methods
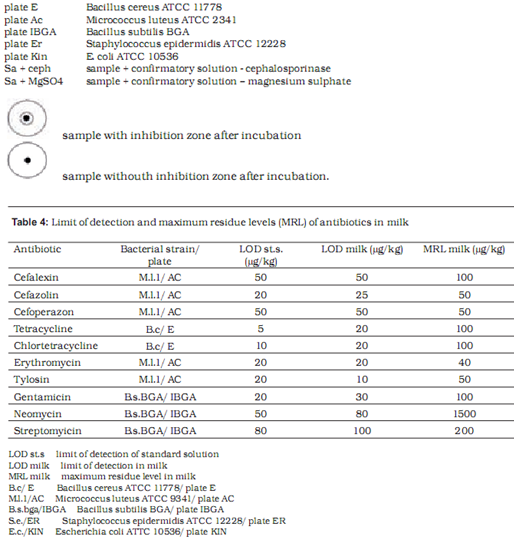
Microbiological methods are based on the measurement and evaluation of zones of inhibited bacterial growth on media. Two test strains are used to assess the presence of each antibiotic – one maximally sensitive and the other resistant to the tested substance. With the combination of different sensitive and resistant bacterial strains, specific antibiotic groups present in the sample can be identified. In our research we used the following strains with previously established sensitivity and resistancy profiles: Bacillus cereus ATCC 11778, Micrococcus luteus ATCC 2341, Escherichia coli ATCC 10536, Staphylococcus epidermidis ATCC 12228 and E. coli ATCC 10536 (manufactured by OXOIDTM).
Preparation of bacterial cultures and media
Bacterial strains stored as cultures in original bacterial loops (Culti loop) were applied to a test tube containing 1ml Trypton soya broth (TSB) medium and incubated at 37 oC for one hour. The culture was then inoculated on blood agar and incubated for further 16 hours at the same temperature. Afterwards the purity of bacterial colonies was assessed with a light microscope and pure colonies were stored in a fridge at temperatures between 2 oC and 8 oC for up to one month. To compose test plates, bacterial culture was diluted with normal saline containing peptone water to produce a suspension which was then incubated at 37 oC for one hour and afterwards added to the agar medium specified below. The suspension density was standardised with the Mc Farland method.
Basic media for preparation of test plates were antibiotic agar No. 1 (MerckTM) and antibiotic agar No. 2 (MerckTM). Antibiotic agar No. 1 was prepared as follows: 1000 ml of distilled water was added to 30, 5 g of the medium, left for 15 min and then heated to boiling point so that the medium was completely dissolved. The medium was then autoclaved at 121 oC for 15 min. For antibiotic agar No. 2 1000 ml of distilled water was added to 15, 5 g of medium and then the same procedure was followed. After autoclaving, the pH of the media was set to desired values: pH 8 for Er, I BGA, Kin and AC plates and pH6 for E plates.
Preparation of test plates
Test plates were marked according to the bacterial strain added to the medium: AC plate – Micrococcus luteus ATCC 2341, ER plate - Staphylococcus epidermidis ATCC 12228, I-BGA plate - Bacillus subtilis BGA, Kin plate - E. coli ATCC 10536 and E plate – Bacillus cereus ATCC 11778. The pH of the medium was maintained at 8.0 for AC, E and ER plates and at 6.0for I-BGA and Kin plates. We defined the tolerance for the width of inhibition zone at (as) 8.5 mm – 0.5 mm wider than the width of the metal cylinder containing the sample. Inhibition zones between 8 mm and 8.5 mm wide were considered a non-specific reaction. To prepare a test plate 0.45 ml of suspension of bacterial culture was added to 40 ml of basic medium and heated to 40 0C. Kin plate was an exception where 0.2 ml of suspension was added to 50 ml of medium. The mixture of medium and bacterial culture was poured into a petri dish (5 ml of mixture into each petri dish). At room temperature the petri dishes with silified medium were enveloped in a parafilm and stored in a fridge. The storage period of test plates was one week. Before application of samples to test plates, plates were warmed at room temperature for 20 to 30 min.
Preparation of milk samples
To test the sensitivity of our method, milk samples containing known concentrations of standard antibiotics were inoculated on test plates. Prior to the addition of antibiotics, milk was always tested for the presence of inhibitory substances.
As the initial step, standard antibiotic solutions were prepared using reference chemical composition and purity. Standard antibiotics in powder were dissolved in appropriate solvents: tetracyclines in phosphate buffer with pH value 4.5, cephalosporines in phosphate buffer with pH value 6.0, aminoglicosides in phosphate buffer with Ph value 8.0, and macrolides in methanol. Standard solutions were diluted to desired concentrations with UHT milk containing 1.6% fat (Ljubljanske mlekarne). These samples of milk with known concentrations of antibiotics were then poured into 10-ml test tubes and heated to 80 oC for 5 min to avoid later non-specific reaction on test plates. After heating, the samples were cooled and transferred to plates in 8 mm wide cylinders. Test plates were incubated at 37 oC (I-BGA, AC, Er, Kin) or at 30 oC (E plate) for 18-24 hours. For each antibiotic we used milk samples containing antibiotic concentrations equal to MRL and half the MRL for that substance. If at half the MRL the result was still positive, lower concentrations of antibiotic were applied until the minimal level of detection was reached.
Confirmation solutions
To confirm the presence of antibiotic groups or their individual representatives we used confirmation solutions. These solutions inhibit the action of certain antibiotics and can help to distinguish between antibiotic groups which cause inhibition zones on the same test plates. Magnesium sulphate (MgSO4) was used to neutralise the aminoglicosides and cephalosporinase enzyme to neutralise the cephalosporines.
25 μl of 20% MgSO4 solution in water was added to the sample on E, AC and I-BGA plates where inhibition zones are produced by aminoglicosides, macrolides or tetracyclines. 25 μl of cephalsporinase was added to samples on AC and I-BGA plates to identify cephalosporines.
Evaluation of results
Results of microbial methods can be evaluated both qualitatively and quantitatively. Qualitative results are obtained by analysing the effect of antibiotics on a combination of sensitive and resistant bacterial strains. When required, neutralizing substances (confirmation solutions) can help to differentiate between antibiotics with similar action on test bacterial strains.
Quantitatively the concentration of antibiotic can be assessed with microbial methods if the sample contains a known antibiotic or an antibiotic that has previously been identified qualitatively. In each case a calibration curve is required.
Results
We have confirmed sensitive and resistant bacterial strains for all antibiotic groups tested in our study. Based on our results we chose to use Bacillus cereus ATCC 11778 (E plate) as the sensitive and Micrococcus luteus ATCC 9341 (AC plate) as the resistant strain for tetracycline and chlortetracycline from the tetracyclines group. For tylosine and erythromycine from the macrolides group Micrococcus luteus ATCC 9341 (AC plate) was chosen as the sensitive and Escherichia coli ATCC10536 (Kin plate) as the resistant strain. For gentamycine, sterptomycine and neomycine from the aminoglicosides group Bacillus subtilis BGA (I-BGA plate) was chosen as the susceptible and Staphylococcus epidermidis ATCC 12228 (ER plate) the resistant strain. For cephalexine, cephoperasone and cephasoline from the sensitive group Micrococcus luteus ATCC 9341 (AC plate) was chosen as the susceptible and Staphylococcus epidermidis ATCC 12228 (ER plate) as the resistant strain.
We differentiated between antibiotic groups using a combination of five test plates (Table 3). To discriminate between aminoglicosides and macrolides we had to utilise used magnesium sulphate which inactivates the aminoglicosides. To discriminate between cephalosporines and macrolides we used the cephalosporinase enzyme.
Table 4 shows the limit of detection for milk samples containing standardised antibiotic solutions on selected test plates. The level of detection was at or below the MRL in all tested antibiotics.
Discussion
Microbial methods for detection of antibiotic residues in food of animal origin are used as a screening method in the majority of laboratories in Europe that deal with analyses of drug residues in food.They are always the method of choice for screening purposes as they allow qualitative detection of antibiotics in the sample and identification of antibiotic groups. This facilitates subsequent confirmation of specific antibiotic residues with chemical methods. Microbial methods are relatively inexpensive, easy to use, do not require expensive equipment and can be efficiently adopted by laboratory staff. Although minimal expenditure is a significant factor of analyses, no test is valuable if it does not give reliable results. We succeeded in developing a microbial method which is sensitive and meets the legislative requirements – to detect concentrations of antibiotics below the MRL. For some antibiotics the level of detection was at half the MRL or lower.Microbial methods are semi quantitative, therefore any positive or suspicious result should be confirmed by chemical methods. In accordance with the EC 2002/657/EC regulation results of microbial methods are not reported as negative and positive, but as satisfactory or suspect when the MRL is exceeded.
Although the STAR five-plate test is the official method approved by the Community Reference Laboratory, many variations of microbial methods are used across the world and most laboratories apply a specific approach with a different number and types of bacterial strains and therefore a different number of test plates.
REFERENCES
1. Indian Pharmacopoeia 2007, Published By The Indian Pharmacopoeia Commission,Government Of India Ministry Of Health & Family Welfare,Central Indian Pharmacopoeial Laboratory,Printed By National Institute Of Science Communication And Information Resources (NISCAIR) Council Of Scientific & Industrial Research, Page No.45
2. The United State Pharmacopoeia,The National Formulary,Published By United States Pharmacopoeial Convention,INC.12601,Twinbrook Parkwag,Rockville,MD 20852,Asian Edition 2005,Page No.2256
3. slovetres.si/files/pdf/volume2006/vol43_4/SlovVetRes_43_4_pp161-168.pdf
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






