

 About Authors:
About Authors:
Nareshkumar. G1*, A.Eashwar rao2, Ch.Ganga prasad3.
1*Center for Pharmaceutical Sciences, IST, JNTU Hyderabad, India,
2Talla Padmavathi College of Pharmacy, Warangal, India,
3National College of Pharmacy, Shimoga, Karnataka, India.
*nareshkumar62@gmail.com
ABSTRACT:
Liquisolid technique is a new and propitious method that can change the dissolution rate of poorly water soluble drugs. According to the new formulation method of liquisolid compacts, liquid medications such as solutions or suspensions of water insoluble drugs in suitable non-volatile liquid vehicles can be converted into acceptably flowing and compressible powders by blending with selected powder excipients. It has been speculated that such systems exhibit enhanced release profiles. In this case, even though the drug is in a solid dosage form, it is held within the powder substrate in solution or, in a solubilized, almost molecularly dispersed state, which contributes to the enhanced drug dissolution properties. Successful liquisolid tablet is a determination of optimal flowable liquid retention.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1595
INTRODUCTION
The active ingredient in a solid dosage form must undergo dissolution before it is available for absorption from the gastrointestinal tract (1, 2). The poor dissolution rates of water-insoluble drugs are still a substantial problem confronting the pharmaceutical industry (3, 4). The absorption rate of a poorly water-soluble drug, formulated as an orally administered solid dosage form, is controlled by its dissolution rate in the fluid present at the absorption site, that is the dissolution rate is often the rate-determining step in drug absorption (1). Over the years, various solid dosage formulation techniques, to enhance the dissolution of poorly soluble substances, have been introduced with different degrees of success. The use of water-soluble salts and polymorphic forms, the formation of water soluble molecular complexes, drug micronization, solid dispersion, coprecipitation, lyophilisation, microencapsulation, and the inclusion of drug solutions or liquid drugs into soft gelatin capsules are some of the major formulation tools that have been shown to enhance the dissolution characteristics of water-insoluble drugs (5). Among them, the technique of liquisolid compacts is one of the most propitious (2–4). Liquisolid compacts are acceptably flowing and compressible powdered forms of liquid medications. The term liquid medication implies oily, liquid drugs and solutions or suspensions of water-insoluble solid drugs carried in suitable nonvolatile solvent systems termed the liquid vehicles. Using this new formulation technique, a liquid medication may be converted into a dry-looking, non-adherent, free-flowing, and readily compressible powder by a simple blending with selected powder excipients referred to as the carrier and coating materials (7–9). Various grades of cellulose, starch, lactose, and so on, may be used as the carriers, whereas very fine-particle-size silica powders may be used as the coating (or covering) materials (10, 11). In liquisolid compacts, even though the drug is in a tableted or encapsulated dosage form, it is held in a solubilized liquid state, which consequently contributes to increased drug wetting properties, thereby enhancing drug dissolution. Another advantage of liquisolid systems is that their production cost is lower than that of soft gelatin capsules because the production of liquisolid systems is similar to that of conventional tablets (7–9).
[adsense:468x15:2204050025]
THEORY OF LIQUID SOLID SYSTEMS
A powder can retain only limited amounts of liquid while maintaining acceptable flow and compression properties. To calculate the required amounts of powder excipients (carrierand coating materials) a mathematical approach for the formulation of liqui-solid systems has been developed by Spireas (12,13). This approach is based on the flowable (Φ-value) and compressible (Ψ-number) liquid retention potential introducing constants for each powder/liquid combination. The Φ-value of a powder represents the maximum amount of a given non-volatile liquid that can be retained inside its bulk [w/w] while maintaining an acceptable flowability. The flowability may be determined from the powder flow or by measurement of the angle of repose. The Ψ-number of a powder is defined as the maximum amount of liquid the powder can retain inside its bulk [w/w] while maintaining acceptable compactability resulting in compacts of sufficient hardness with no liquid leaking out during compression14. The compactability may be determined by the so-called “pactisity” which describes the maximum (plateau) crushing strength of a one-gram tablet compacted at sufficiently high compression forces. The terms “acceptable flow and compression properties” imply the desired and thus preselected flow and compaction properties which must be met by the final liqui-solid formulation.
Depending on the excipient ratio (R) of the powder substrate an acceptably flowing and compressible liqui-solid system can be obtained only if a maximum liquid load on the carrier material is not exceeded. This liquid/carrier ratio is termed “liquid load factor Lf [w/w] and is defined as the weight ratio of the liquid formulation (W) and the carrier material (Q) in the system:
Lf = W/Q------ (1)
‘R’ represents the ratio between the weights of the carrier (Q) and the coating (q) material present in the formulation:
R =Q/q------ (2)
The liquid load factor that ensures acceptable flowability(Lf) can be determined by:
Lf =Φ+ φ. (1/R) ----- (3)
Where Φ and φ are the Φ-values of the carrier and coating material, respectively. Similarly, the liquid load factor for production of liqui-solid systems with acceptable compactability (ΨLf) can be determined by:
ΨLf= Ψ + ψ.(1/R) ------- (4)
Where Ψ and ψ are the Ψ-numbers of the carrier and coating material, respectively. In Table-1 examples of liqui-solid formulation parameters of various powder excipients with commonly used liquid vehicles are listed.
Table 1: Liqui-solid formulation parameters of various powder excipients with commonly used liquid vehicles Powder.
|
Excipient or System |
Φ-values |
Φ-values |
||
|
Propylene glycol |
PEG-400 |
Propylene glycol |
PEG-400 |
|
|
Avicel pH 102 |
0.16 |
0.005 |
0.224 |
0.242 |
|
Avicel pH 200 |
0.26 |
0.02 |
0.209 |
0.232 |
|
Cab-O-Sil M5(silica)* With Avicel pH 102 |
3.31 |
3.26 |
0.560 |
0.653 |
|
Cab-OSilM5(silica)* With Avicel pH 200 |
2.57 |
2.44 |
0.712 |
0.717 |
*Included as coating material in carrier/coating powder systems.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Therefore, the optimum liquid load factor (Lo) required to obtain acceptably flowing and compressible liqui-solid systems are equal to either ΦLforΨLf, whichever represents the lower value.
As soon as the optimum liquid load factor is determined, the appropriate quantities of carrier (Qo) and coating (qo) material required to convert a given amount of liquid formulation (W) into an acceptably flowing and compressible liqui-solid system may be calculated as follows:
Q0 = W/Lo-----(5) And q0 = Q0/R-------(6)
The validity and applicability of the above mentioned principles have been tested and verified by producing liquisolid compacts possessing acceptable flow and compaction properties12.
MECHANISMS OF ENHANCED DRUG RELEASE FROM LIQUID-SOLID SYSTEMS
Several mechanisms of enhanced drug release have been postulated for liqui-solid systems. The three main suggested mechanisms include an increased surface area of drug available for release, an increased aqueous solubility of the drug, and an improved wettability of the drug particles. Formation of a complex between the drug and excipients or any changes in crystallinity of the drug could be ruled out using DSC and XRPD measurements14.
a. Increased Drug Surface Area
If the drug within the liqui-solid system is completely dissolved in the liquid vehicle it is located in the powder substrate still in a solubilized, molecularly dispersed state. Therefore, the surface area of drug available for release is much greater than that of drug particles within directly compressed tablets13.
b. Increased Aqueous Solubility of the Drug
In addition to the first mechanism of drug release enhancement it is expected thatCs, the solubility of the drug, might be increased with liqui-solid systems. In fact, the relatively small amount of liquid vehicle in a liqui-solid compact is not sufficient to increase the overall solubility of the drug in the aqueous dissolution medium. However, at the solid/liquid interface between an individual liqui-solid primary particle and the release medium it is possible that in this microenvironment the amount of liquid vehicle diffusing out of a single liqui-solid particle together with thedrug molecules might be sufficient to increase the aqueous solubility of the drug if the liquid vehicle acts as a cosolvent13.
c. Improved Wetting Properties
Due to the fact that the liquid vehicle can either act as surface active agent or has a low surface tension, wetting of the liqui-solid primary particles is improved. Wettability of these systems has been demonstrated by measurement of contact angles and water rising times15.Many poorly soluble drugs have been formulated as liquisolid systems showing enhanced drug release. Different liquid vehicles, carrier and coating materials were used to formulate these drug delivery systems.
ADVANTAGES OF LIQUID-SOLID COMPACT
· Several slightly and very slightly water-soluble and practically water-insoluble liquid and solid drugs can be formulated into liqui-solid systems.
· Even though the drug is in a tablet or capsuleform, it is held in a solubilized liquid state, which contributes to increased drug wetting properties, thereby enhancing drug dissolution.
· Production cost is lower than soft gelatin capsules21.
· This technique is successfully applied for low dose water insoluble drug.
· The absolute bioavability of the drug from the liquisolid tablet is 15% higher than that commercial one.
· Their production cost is lower than that of soft gelatin capsules because the production of liquisolid systems is similar to that of conventional tablets.
· Drug dissolution from liquisolid compact is independent to the volume of dissolution media.
· Most of liquid or solid ‘water insoluble drug’ may be formulated into immediate release or sustained release ‘Liquisolid compact’ or ‘Liquisolid microsystem.
LIMITATIONS
· Not applicable for the formulation of high dose insoluble drugs.
· If more amount of carrier is added to produce freeflowing powder, the tablet weight increases to more than one gram which is difficult to swallow.
· Acceptablecompression properties may notbeachieved since during compression liquid drug may be squeezed out of the liquid-solid tablet resulting in tablets of unsatisfactory hardness21.
· Introduction of this method on industrial scale and to overcome the problems of mixing small quantities of viscous liquid solutions onto large amounts of carrier material may not be feasible.
APPLICATIONS OF LIQUID-SOLID COMPACTS
Following are few important applications of liquid-solid compacts16-20:
· Rapid release rates are obtained in liqui-solid formulations
· These can be efficiently used for water insoluble solid drugs or liquid lipophilic drugs.
· Sustained release of drugs which are water soluble drugs such as propranolol hydrochloride has been obtained by the use of this technique.
· Solubility and dissolution enhancement.
· Designing of controlled release tablets.
· Application in probiotics.
CLASSIFICATION
A. Based on the type of liquid medication contained therein, liquisolid systems may be classified into three subgroups:
1. Powdered drug solutions
2. Powdered drug suspensions
3. Powdered liquid drugs
The first two may be produced from the conversion of drug solutions or (e.g. prednisolone solution in propylene glycol) or drug suspensions (e.g. gemfibrozil suspension in Polysorbate 80), and the latter from the formulation of liquid drugs (e.g. clofibrate, valproic acid, liquid vitamins, etc.), into liquisolid systems.
B. Based on the formulation technique used, liquisolid systems may be classified into two categories, namely,
1. Liquisolid compacts
2. Liquisolid microsystems
Liquisolid compacts are prepared using the previously outlined method to produce tablets or capsules, whereas the liquisolid microsystems are based on a new concept which to produce an acceptably flowing admixture for encapsulations22.
PREPARATION OF LIQUID SOLID COMPACTS
As shown in figure, a liquid lipophilic drug (e.g.Chlorpheniramine, Clofibrate, etc.) can be converted into a liqui-solid system without being further modified. On the other hand, if a solid water-insoluble drug (e.g. Hydrochlorothiazide, Prednisone, etc.) is formulated, it should be initially dissolved or suspended in a suitable nonvolatile solvent system to produce a drug solution or drug suspension of desired concentration.
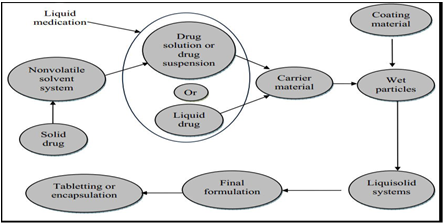
Fig.1: Steps involved in the preparation of liquid solid systems
Next, a certain amount of the prepared drug solution or suspension, or the liquid drug itself, is incorporated into a specific quantity of carrier material which should be preferably of a porous nature and possessing sufficient absorption properties, such as powder and granular grades of microcrystalline and amorphous cellulose are most preferred as carriers. The resulting wet mixture is then converted into a dry-looking, non-adherent, free-flowing and readily compressible powder by the simple addition and mixing of a calculated amount of coating material. Excipients possessing fine and highly adsorptive particles, such as various types of amorphous silicon dioxide (silica), are most suitable for this step. Before compression or encapsulation, various adjuvants such as lubricants and disintegrates (immediate) or binders (sustained-release) may be mixed with the finished liqui-solid systems to produce liqui-solid compacts i.e. tablets or capsules,23,24,25 .
COMPONENTS OF LIQUISOLID COMPACT FORMULATION
Liqui-solid compact mainly includes
1. Non volatile solvent
2. Disintegran
3. Carrier material
4. Coating material
1. Nonvolatile Solvent
Nonvolatile Solvent should be Inert, high boiling point, preferably water-miscible and not highly viscous organic solvent systems and compatible with having ability to solubilize the drug. The non-volatile solvent acts as a binding agent in the liquisolid formulation various non-volatile solvents used for the formulation of liquisolid systems include Polyethylene glycol 200 and 400, glycerin, polysorbate 80 and propylene glycol.
2. Disintegrant
Super disintigrants increases the rate of drug release, water solubility and wettability of liquisolid granules. Mostly superdisintigrantslike sodium starch glycolate and crosspovidone.
3. Carrier Materials
Carrier material should be porous material possessing sufficient absorption properties which contributes in liquid absorption. The carrier and coating materials can retain only certain amounts of liquid and at the same time maintain acceptable flow and compression properties hence; increasing moisture content of carrier’s results in decreased powder flowability. These include grades of microcrystalline cellulose such as avicel PH 102 and avicel PH 200.
4. Coating Materials
Coating material should be a material possessing fine and highly adsorptive particles which contributes in covering the wet carrier particles and displaying a dry looking powder by adsorbing any excess liquid. Coating material is required to cover the surface and maintain the powder flowability34.Coating material includes silica (Cab-O-Sil) M520,Aerosil 200, Syloid-244FP etc.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Examples of drugs that can be incorporated into liquisolid systems22, 23:
· Chlorpheniramine
· Digoxin
· Nifedipine
· Clofibrate
· Gemfibrozil
· Etoposide
· Carbamazepine
· Hydrochlorothiazide
· Methyclothiazide
· Spironolactone
· Hydrocortisone
· Piroxicam
· Indomethacin
· Ibuprofen
PRE-COMPRESSION STUDIES OF THE LIQUID SOLID SYSTEM
Flow Properties of the Liqui-Solid System
The flow properties of the liqui-solid systems were estimated by determining the angle of repose, Carr’sindex, and Hausner’s ratio. The angle of repose was measured by the fixed funnel and freestanding cone method. The Bulk density and Tap densities were determined for the calculation of Hausner’s ratio andCarr’s Index.
Angle of repose
The angle of repose physical mixtures of liqui-solid compacts were determined by fixed funnel method. The accurately weighed physical mixtures of liqui-solid compacts were taken in a funnel. The height of the funnel was adjusted in such a way that the tip of the funnel just touches the apex of the heap of the powder. The powder was allowed to flow through the funnel freely into the surface. The height and diameter of the powder cone was measured and angle of repose was calculated.
Tan θ= h/r
Where, θ is the angle of repose h is the height in cms, r is the radius in cms, Values for angle of repose ≤ 30 usually indicate a free flowing material and angles ≥ 400 suggest a poorly flowing material. 25- 30 showing excellent flow properties, showing good flow properties, 36-40 showing fair flow properties, 41-45 showing passable flow properties.
Bulk Density
The loose bulk density and tapped density were determined by using bulk density apparatus. Apparent bulk density was determined by pouring the blend into a graduated cylinder. The bulk volume (Vb) and weight of the powder (M) was determined. The bulk density was calculated using the formula:
Db=M/Vb
Where, M is the mass of powder and Vb is bulk volume of powder.
Tapped Density
The measuring cylinder containing a known mass of blend was tapped for a fixed time. The minimum volume (Vt) occupied in the cylinder and the weight (M) of the blend was measured. The tapped density was calculated using the formula:
Dt= M/Vt
Where, M is the mass of powder and Vt is tapped volume of powder.
Carr’s Index (%)
The compressibility index has been proposed as an indirect measure of bulk density, size and shape, surface area, moisture content and cohesiveness of material because all of these can influence the observed compressibility index. The simplest way for measurement of free flow of powder is Carr’s Index, an indication of the ease with which a material can be induced to flow is given by Carr’s index (CI) which is calculated as follows:
CI (%) = [(Tapped density – Bulk density) / Tapped density] x 100
The value below 15% indicates a powder with usually gives rise to good flow characteristics, whereas above 25% indicates poor flowability. 1-10 showing excellent flow properties, 11-25 showing good flow properties 16-20, showing fair to passable, 21-25 showing passable.
Hausner’s Ratio
Hausner’s ratio is an indirect index of ease of powder flow. It is calculated by the following formula.
Hausner’s Ratio=Tapped density (ρt) / Bulk density (ρb)
Where ρt is tapped density and ρb is bulk density. Lower Hausner’s ratio (<1.25) indicates better flow properties than higher ones, between 1.25 to 1.5 showing moderate flow properties and more than 1.5 poor flow.
POST COMPRESSION STUDIES OF LIQUI-SOLID COMPACTS
Weight Variation
Twenty tablets were randomly selected from each batch and individually weighed. The average weight and standard deviation three batches were calculated. It passes the test for weight variation test if not more than two of the individual tablet weights deviate from the average weight by more than the allowed percentage deviation and none deviate by more than twice the percentage shown. It was calculated on an electronic weighing balance.
Thickness
The thickness of liqui-solid tablets was determined by using Digital micrometer. Ten individual tablets from each batch were used and the results averaged.
Hardness
The hardness of the tablets was determined by using Monsanto hardness tester. Five individual tablets from each batch were and results averaged.
Friability
The friability values of the tablets were determined using a Roche-type friabilator. Accurately weighed six tablets were placed in Roche friabilator and rotated at 25 rpm for 4 min. Percentage friability was calculated using the following equation.
Friability = ([WO – W] /WO) * 100
Where,
WO = Weight of the tablet at time zero before revolution.
W = Weight of the tablet after 100 revolutions.
Disintegration Test
Six tablets were taken randomly from each batch and placed in USP disintegration apparatus baskets Apparatus was run for 10 minutes and the basket was lift from the fluid, observe whether all of the tablets have disintegrated.
In-vitro Release
Drug release from liqui-solid tablets was determined by using dissolution test United States Pharmacopoeia (USP) 24 type II (paddle). 5ml aliquots of dissolution media were withdrawn each time at suitable time intervals (5, 10, 15, 20, 25, 30, 45 and 60 minutes.) and replaced with fresh medium. After withdrawing, samples were filtered and analyzed after appropriate dilution by appropriate analytical method. The concentration was calculated using standard calibration curve.
REFERENCES:
1. Kim KH and Singh BN. Drug delivery–oral route, In: Swarbrick J, Boylen JC, Encyclopedia of pharmaceutical technology,2nd Ed, New York: Marcel Dekker Inc,2002;886-890.
2. Allen LV, Popvich NV and Ansel HC. Tablets In: Ansel’s pharmaceutical dosage forms and drug delivery systems, 8 th Ed; India, B. I. publications pvt.ltd. 227-259.
3. Rudnic EM and Kottke MK. Tablet dosage forms, In: Banker GS, Rhodes CT. Modern pharmaceutics, 4thed, New York ,Marcel Dekker Inc, 2002; 333-394.
4. Alderborn G. Tablet and compaction In: Aulton M. Pharmaceutics: The science of dosage form design, 2nd Ed, Churchill Livingstone, Longman group, Edinburgh, 2002;114-138.
5. Baboota S, Ali J and Ahuja A. Tablet, Available at http://www.pharmpedia.com/Tablet.
6. Charman SA and Charman WN. Oral modified release delivery systems, In: Rathbone MJ, Hadgraftb J, Roberts MS. Modified release drug delivery technology, New York, Marcel Dekker Inc. 2003;313-320.
7. Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J PharmacolToxicol Methods, 2000;44:235-249.
8. Amidon GL, Lennernas H and Crison JR. A theoretical basis for biopharmaceutics drugs classification: the correlation of in-vitro drug product dissolution and in-vivo bioavailability. Pharm Res. 1999;12: 413-420.
9. Vikas A Saharan andVipinKumar.Dissolution Enhancement of Drugs. PartI:Technologies and Effect of Carriers, International Journal of Health Research, June2009;2(2):107-124.
10. NeelamSeedher and SonuBhatia.Solubility Enhancement of Cox-2 Inhibitors Using Various Solvent Systems AAPS PharmSciTech.2003;4 (3)33.
11. Fahmy RH and KassemMA.Enhancement of famotidine dissolution rate through liquisolid tablet formulation: in vitro and in vivo evaluation, Eur J Pharm Biopharm, 2008;69(3):993-1003.
12. Spireas, S. Liqui-solid systems and methods of preparing same. U.S. Patent 6423339B1 (2002)
13. Spireas, S., Sadu, S.Enhancement of prednisolone dissolution properties using liqui- Solid compacts.Int. J. Pharm. 1998,166: 177-188.
14. Spireas, S., Sadu, S. Enhancement of prednisolone dissolution properties using liqui-solid compacts. Int. J. Pharm. 1998, 166: 177-188.
15. Yadav, V.B., Nighute, A.B., Yadav, A.V., Bhise, S.B. Aceclofenac size enlargement by non-aqueous granulation with improved solubility and dissolution. Arch. Pharm. Sci. and Res. 2009,1: 115-122.
16. Kulkarni Ajit S., AloorkarNagesh H., Mane Madhav S. and GajaJayashree B., Liqui-solid Systems. International Journal of Pharmaceutical Sciences and Nanotechnology,2010,3(1),795-802.
17. Bindu MB, Kusum B and David Banji, Novel Strategies for Poorly Water Soluble Drugs, International Journal of Pharmaceutical Sciences Review and Research,2010,4(3),1-5.
18. Sharma, A., Jain, C.P. Techniques to enhance solubility of poorly soluble drugs: a review. J.Global Pharm. Tech.2010,2: 18-28.
19. Saharan, V.A.,Kukkar, V., Kataria, M., Gera, M., Choudhury, P.K. Dissolution enhancement of drugs. Part I: technologies and effect of carriers. Int. J. Health Res. 2009,2: 107-124.
20. Saharan, V.A.,Kukkar, V., Kataria, M., Gera, M., Choudhury, P.K. Dissolution enhancement of drugs. Part II: effect of carriers. Int. J. Health Res. 2009,2: 207-223.
21.S.Spireas, US Patent, US 6,423,339 B1.
22. Nokhodchi A. The effect of type and concentration of vehicles on the dissolution rate of a poorly soluble drug (indomethacin) from liquisolid compacts. J pharm Sci. 8(1):18-25.
23.SpirasS.Liqui-solid systems and methods for preparing same, United States patent6, 423, 339 B1, (2002).
24.Spiras S, Bolton SM. Liqui-solid systems and methods for preparing same, United States patent 6,096,337, 2000.
25.Spiras S, Wang T, Grover R. Effect of powder substrate on dissolution properties of methyl clorthiazideLiqui-solid compacts, Drug. Dev. Ind. Pharm. 1999,25: 63-168.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






