

 About Authors:
About Authors:
Dr.Pushpendra Kumar Tripathi(director of RITM(pharmacy), *Shipra srivastava
Rameshwaram Institute Of Technology And Management
Lucknow.
*shipra.hanny1987@gmail.com
Abstract:
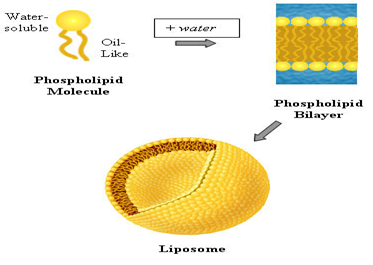
The goal of any drug delivery system is spatial placement and temporal delivery of the medicament. Research works are going on to prepare an ideal drug delivery system which satisfies these needs. Researches carried out by ALEC BINGHAM lead to the development of a new drug delivery system called as liposome. Liposomes are small vesicles (_100 nm) composed of various of lipid molecules which build their membrane bilayers. These formulations are mainly composed of phosphatidylcholine (PC) and other constituents such as cholesterol and lipid-conjugated hydrophilic polymers . Liposomes are biodegradable and biocompatible in nature.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1348
introduction
LIPOSOMES
Liposomes are simple microscopic vesicles in which an aqueous volume is entirely enclosed by membrane composed of lipid molecules. Various amphipathic molecules have been used to form liposomes(1,2)
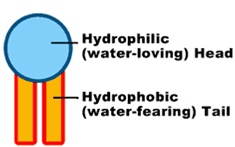
Single phospholipid with it's hydrophilic head and hydrophobic tail
Phospholipids are the major structural components of the biological membranes in the human body, where two types of phospholipids exist i.e. phosphodiglycerides and sphingolipids, together with their corresponding hydrolysis products. The way they work and form membranes are elegant and miraculous. Each phospholipid molecule has three major parts, one head and two tails. The head is made from three molecularcomponents: choline, phosphate, and glycerol. The head is hydrophilic— in otherwords, it is attracted to water. Each tail is a long, essential fatty acid chain. These fattyacids are hydrophobic— that is, they are repelled by water. The drug molecules can either be encapsulated in aqueous space or intercalated in to the lipid bilayers.
The number of components of the liposomes is varied; however phospholipids and cholesterol are the main components. The most commonly used phospholipids include phosphatidyl choline (PC). PC is an amphipathic molecule in which a glycerol bridge links a pair of hydrophobic acyl hydrocarbon chains, with a hydrophilic polar head group, phospho choline. Phosphatidyl choline, also known as “lecithin”, can be derived from natural and synthetic sources(3,4,5,6,7)
[adsense:468x15:2204050025]
Generally phospholipid are represented as follows:

*Molecules of pc are not soluble in water.
*In aqueous media they align themselves closely in planner bilayers sheets in order to minimize the unfavorable action between the bulk aqueous phase and the long hydrocarbon fatty chain (i.e they orient themselves so that the fatty acid chains face each other and the polor heads face the aqueous phase –this reduces the instability which exists when the molecule exist alon
*Such unfavorable interactions are completely eliminated when the sheets fold on themselves to form closed sealed vesicle.
In short this is what happens:
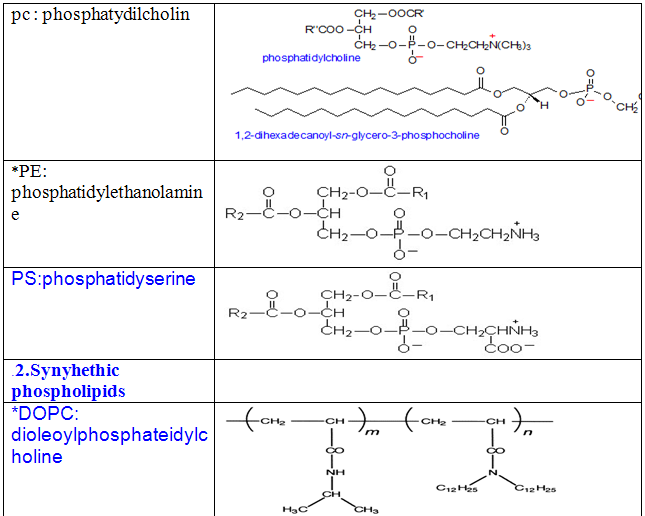
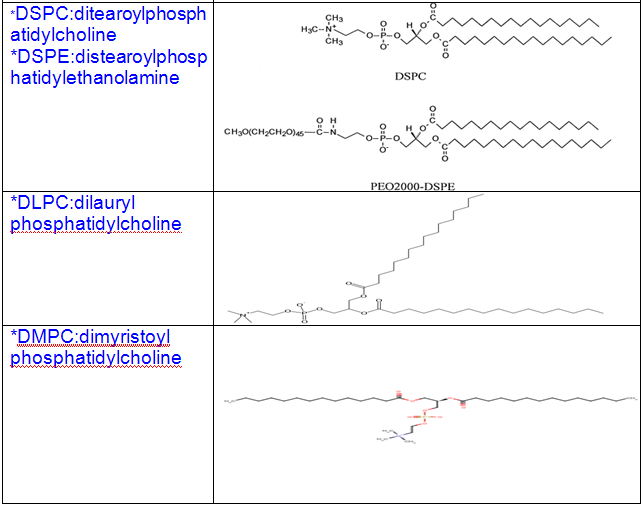
Some other common phospholipids:
1.Naturally ocuring phospholipids:
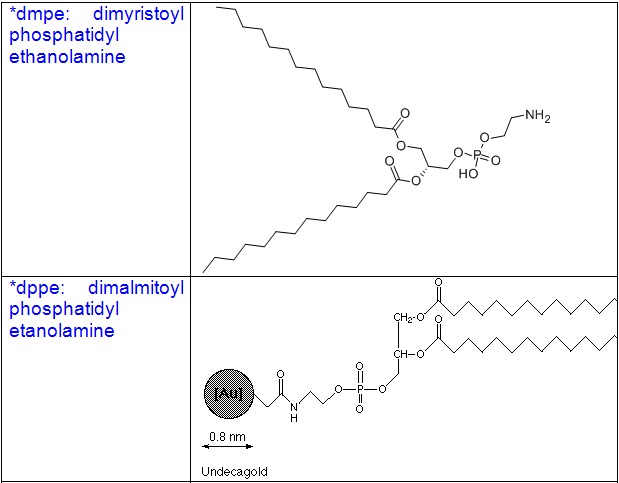
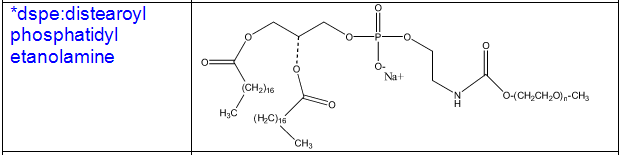
Cholesterol
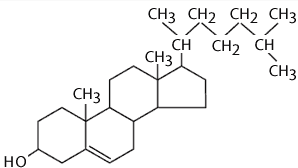
*Incorporation of sterols in liposome bilayer brings about major changes in preparation of these membrane.
*cholesterol by itself does not forms a bilayer structure .
*however cholesterol acts fluidity buffer i.e below the phase transition temperature it make the membrane less ordered and slightly more permeable while above the phase transition temperature it makes the membrane more odered and stable.
*it can be incorporated into phospholipid membrane in very high concentration upto 1:1 or even 2:1 molar ratio of cholesterol to pc.
*cholesterol insert into the membrane with its hydroxyl groups oriented towards the aqueous surface and aliphatic chain aligned parallel to the acyl chain in the center of the bilayer.
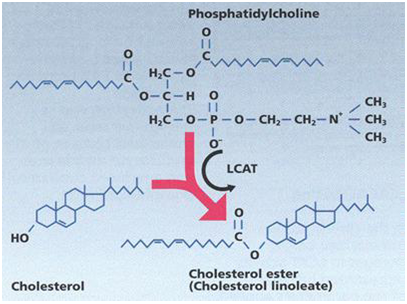
Mechanism of cholesterol acting as a fluidity buffer
*cholesterol incorporation increases the separation between the choline head groups and eliminates the normal electrostatic and hydrogen binding intractions thus pushing the phospholipids apart making the layer less ordered at lower temperature.
*However ,in the higher concentration that cholesterol is used ,the membrane area occupied by the combination of acyl chains and cholestrolis greater than that taken by phosphacholin head group .this differences in area retards chain tilt (the phenomena responsible for phase transition –i.e trans to gauche conformation changes ).above the transition temperature ,the reduction in the freedom of the acyl chains causes the membrane to remain condenced and rigiged with a reduction in area through closer packing and resultant decrease in fluidity(8,9,10,11,12).
Phosphatidylcholine and cholesterol interaction
History of liposome:
*The story of success of liposomes was initiated by Bangham and his colleagues in the early 1960s who observed that smears of egg lecithin reacted with water to form quite intricate structures. They were analyzed by electron microscopy showing that a multitude of vesicles were formed spontaneously. These more or less homogenous lipid vesicles were first called smectic mesophases . Later on, a colleague of Bangham termed them—more euphoniously—liposomes .
* The physiochemical characterization of liposomes had been carried out in 1968-75. Moreover, thin lipid film hydration method had been developed to prepare multilamellar vesicles (MLVs). (13,14). Liposomes were widely used to study the nature of biological membrane because of close resemblance of bilayered membrane with the biological membrane.
*During the late 1970s and early ‘80s, liposomes were re-engineered to maintain their stability so they could circulate in the blood for longer periods of time. While this was accomplished and stealth™ liposomes – ideal for delivering pharmaceutical drugs directly to cells - were developed, theyremained very difficult to produce on a large scale.
* In 1975 – 85Liposome’s utility was improved following basic research that increased the understanding of their stability and interaction characteristic within the system (15). This period also dealt with the discovery of various alternative methods for the preparation of liposomes. Also, due to the availability of vast knowledge about the physio-chemical properties of liposomes, their behavior within the body, their interaction with the cells, attempts had been made to improve their performance as drug carrier systems (16,17,18).
*The development of liposomal drugs with clinical utilityrelied on the development of techniques, which allowed the rapid generation of homogeneous small liposomes and efficient accumulation of drugs into liposomes. This was made possible by the extrusion technique and the pH gradient loading techniques, which were developed in the late 1980s and early 1990s. The first liposomal drug formulation on the US market was the anticancer drug doxorubicin encapsulated in sterically stabilised liposomes (Doxil®). Doxil® was approved by the FDA in 1995. It should be noted that it can take between 5 - 10 years and 50 - 100 million US dollars to bring a liposomal drug from the research and development stage to the market.
*Today, liposomes are used successfully in various scientific disciplines, including mathematics and theoretical physics (topology of two-dimensional surfaces floating in a three dimensional continuum), biophysics (properties of cell membranes and channels), chemistry (catalysis, energy conversion, photosynthesis), colloid science (stability, thermodynamic of finite systems), biochemistry (function of membrane proteins) and biology (excretion, cell function, trafficking and signaling, gene delivery and function). AmbisomeTM, a parenteral amphotericin-B based liposomal product was first in the race, followed by number of other products which are either at the stage of clinical trials or are already in the market . Moreover, renaissance in the liposome research is promising many more products to come in the near future( 18).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Classification of liposome:
Liposomes are spherical, self-closed structures formed by one or several concentric lipid bilayers with an aqueous phase inside and between the lipid bilayers. There are a number of different types of liposomal vesicle:
Based on structural parameter:
1.Multilamellar vesicles: these range in size from 500 to 5,000 nm and consist of several concentric bilayers.
2. Small unilamellar vesicles: around 100 nm in size and formed by a single bilayer.
3. Large unilamellar vesicles: range in size from 200 to 800 nm.
Classification of commonly known lipid vesicles according to their structures and/or preparation:
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Mechanism of action of liposome:
Liposomes as drug delivery systems can offer several advantages over conventional dosage forms especially for parenteral (i.e. local or systemic injection or infusion), topical, and pulmonary route of administration. The preceding discussion shows that liposomes exhibit different biodistribution and pharmacokinetics than free drug molecules. In several cases this can be used to improve the therapeutic efficacy of the encapsulated drug molecules. The limitations can be reduced bioavailability of the drug, saturation of the cells of the immune system with lipids and potentially increased toxicity of some drugs due to their increased interactions with particular cells. The benefits of drug loaden liposomes, which can be applied as (colloidal) solution, aerosol, or in (semi) solid forms, such as creams and gels, can be summarized into seven categories:
(i)Improved solubility of lipophilic and amphiphilic drugs. Examples include Porphyrins, Amphotericin B, Minoxidil, some peptides, and anthracyclines, respectively; furthermore, in some cases hydrophilic drugs, such as anticancer agent Doxorubicin or Acyclovir can be encapsulateded in the liposome interior at concentrations several fold above their aqueous solubility. This is possible due to precipitation of the drug or gel formation inside the liposome with appropriate substances encapsulated (45).
(ii)Passive targeting to the cells of the immune system, especially cells of the
mononuclear phagocytic system (in older literature reticuloendothelial system).
Examples are antimonials, Amphotericin B, porphyrins and also vaccines,
immunomodulators or (immuno)supressors)(45).
(iii)Sustained release system of systemically or locally administered liposomes.
Examples are doxorubicin, cytosine arabinose, cortisones, biological proteins
or peptides such as vasopressin(46).
(iv)Site-avoidance mechanism: liposomes do not dispose in certain organs, such as heart, kidneys, brain, and nervous system and this reduces cardio-, nephro-, and neuro-toxicity. Typical examples are reduced nephrotoxicity of Amphotericin B, and reduced cardiotoxicity of Doxorubicin liposomes(47).
(v)Site specific targeting: in certain cases liposomes with surface attached ligands can bind to target cells (‘key and lock’ mechanism), or can be delivered into the target tissue by local anatomical conditions such as leaky and badly formed blood vessels, their basal lamina, and capillaries. Examples include anticancer, antiinfection and antiinflammatory drugs(48).
(vi)Improved transfer of hydrophilic, charged molecules such as chelators, antibiotics(48)
(vii)Improved penetration into tissues, especially in the case of dermally(48).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Liposome in vivo:
Over the last 30 years much information has been gained concerning the behaviour of liposomes in vivo:
* It was found that clearance of liposomes from the circulation and their biodistribution depend on the physicochemical properties of the liposomes such as liposome size, surface charge and bilayer packing, as wellas on other factors such as dose and route of administration (49,50,51).
*.In order to transport drugs to or into tumour cells liposomes must avoid interactions with circulating cells and proteins in the blood, and uptake by phagocytic cells,which are responsible for their removal from the circulation.
Then they must leave the vasculature (extravasate)at the site of tumour growth.
*Liposomes have then to cross the space between the vasculature and the tumour (interstitial space) and enter the tumour mass. There, dependent on the drug being delivered,the liposomes have to be taken up into the tumour cells and facilitate the delivery of the drug to its intracellular site of action( 52,53)
* For conventional drugs there is no absolute need for the liposomes to associate with the tumour cells and to be taken up into the cells.
Drug encapsulation:
*Spherical phospolipid-based liposomes can be used to carry large amount of small water-soluble molecules (therapeutic drugs) in their aqueous core or lipophilic ones in their lipid bilayer membrane through co-valent, non-covalent or avidin-biotin binding (54).
The active delivery of therapeutics to targeted sites (disease sites) of the body is largely dependent on sufficient local concentration of a therapeutic agent.
Resistance to the rapid clearance by the reticuloendothelial system9, amount of polyethylene glycol(PEG) moiety10 incorporated into the liposome bilayer and adequate amount of transport agents when crossing the endothelial barriers through various mechanisms (e.g. the transport via receptor-mediaton) (55,56) .PEG molecules, which are used as a spacer (_gure 3), results in a better exibility of the targeting vector bound.
That particular binding increases the efficiency of targeting to specice receptors or in general transport of drugs into targeting tissues [10]. The size of liposomes has also been shown to be important factor in the efficient delivery of therapeutic drugs to the disease sites (e.g. antitumor drugs to a tumor site) (57).
Figure { Shematic representation of drug encapsulation in liposomes. Hydrophilic drugs (yellow) are encapsulated in the interrior of the liposome, while hydrophobic ones (violet) are bound in the interior of phospholipid membrane. Vesicles (red), bound on PEG molecules (blue) represent targeting vectors which bind to a special_ cell receptors( 57).
Fig-Liposome-cell interaction. The _first process is liposomal adsorbtion to the cell membrane (lower left). Some of the contents may be released into the extracellular uid and some fraction may pass through the membrane. The second possible process is the uptake of liposomes by endocytosis (upper left), which then degrade in organelles called lysosomes and release the content into the cytoplasm. The third possible process is fusion, where the liposome's content enters the cytoplas directly (upper right). The last mechanism is the exchange of lipids (lower right) .
Encapsulation of drugs in liposomes has several advantages:
*Stable encapsulation of drugs in liposomes changes the drugelimination characteristics (pharmacokinetics) and biodistribution.For example, free drugs injected into the blood stream usually have a large volume of distribution and as a consequence exhibit significant toxicity for healthy tissues.
* Encapsulation of drugs in liposomes can reduce the volume of distribution and decrease toxic side effects in healthy tissues. Furthermore, increased circulation life times result in higher levels of accumulation at disease sites as compared to free drug.
*This can result in increased efficacy if the drug is bioavailable (released from the liposomes)(57,58)
Two problems become immediately obvious when trying to encapsulate drugs into liposomes.
* First, encapsulation becomes more difficult and inefficient as the size of the drug increases. For example, the longest dimension of a 4490 bp plasmid is between 300 - 500 nm and exceeds the diameter of a 100 nm liposome
* Second, the encapsulation efficiencies and drug-to-lipid ratios achieved by ‘passive’ encapsulation techniques such as lipid film hydration are low(57,58)
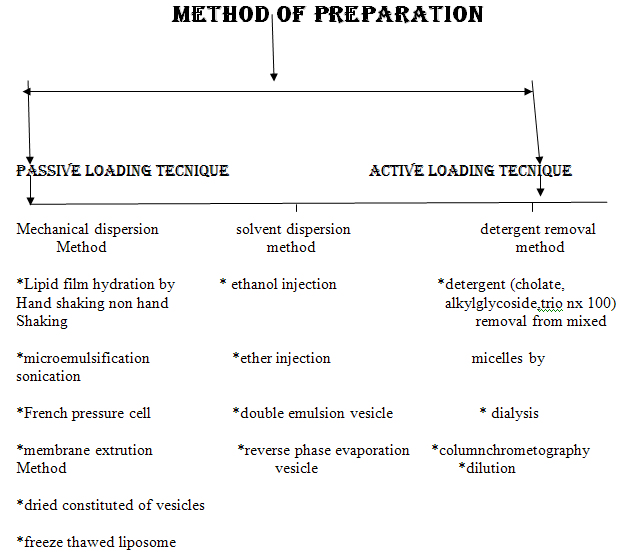
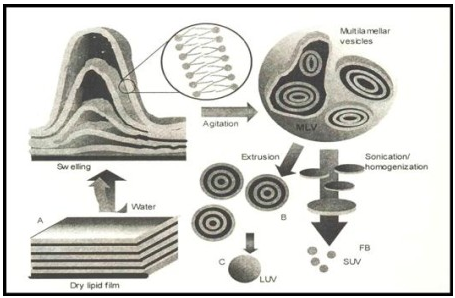
Simplest and most widely used method of Physical dispersion:
Hand shaking method:
- dissolution of the lipid mixure and charge components in chloroform :methanol solvent
- evaporation of the solvent in rotatry evaporator or by hand shaking to form a film
- further dryning of the film by attaching the flask to the manifold of the lyopholizer
- casted film is then dispersed in an aqueous medium.
- upon hydration,lipid swell and peel off the wall of the flask and vesiculate forming multilameller vesicels
Non hand shaken method:
*solution of lipid in chloroform :methenol mixture is spread over the flat bottom conical flask
*the solution is evaporated at room temprature by flow of nitrogen through the flask without disturbing the solution.
*after drying water saturated nitrogen is passed through the flask untill the opacity of the dried lipid film disappears(15-20min)
*after hydration ,lipid is swelled by addition of bulk fluid .the flask is inclined to one side ,10-20 ml of 0.2m sucrose in distilled water (degassed)is introdused down the side of the flask,and the flask is slowly returned to upright orientation .
*the fluid is allowed to run gently over the lipid layer on the bottom of the flask.
*the flask is flushed with nitrogen ,seald and allowed to stand for 2 hrs at 37 degree celceius .take care not distrub the flask in any way.
*after swelling the vesicle are harvested by swirling the contents of the flask gently to yield a milky suspention
Proliposome:
*Method devised to increase the surface of dry lipid while keeping the low aqueous volume.
*in this method ,the lipid are dried down to afinely divided particulate support such as powdered sodium chloride or sorbital or other polysaccharide to give pro liposome
* the lipid are swelled upon adding water to dried lipid coated powder (pro liposome) where the support rapidly dissolves to give a suspension of mlvs in aqueous solution.
*the size of the carrier influences the size and heterogeneity of the liposome
*the method overcomes the problems encountered when storing liposomes themselves in either liquid ,dry or frozen from and is ideally suited for preparation where the material to be entrapped incorporates into lipid membrane.
*in case where 100% entrapement of aqueous component is not essential ,this method is also of value
*for preparing proliposome a special equipment i.e buchi rotatry evaporator’ R’ with water cooled condenser coil and a stainless steel coverd thermocouple connected to a digital thermocouple is required.
*the end of the glass solvent inlet tube is modified to a fine point ,so that the solvent is introduced into the flask as a fine spray.
Method of preparation of proliposome:
*The lipid solution in chloroform (60 mg per ml) is prepared and sorbital powder is introduced into 100 ml flask.
*the flask is then fitted into the evaporate and rotated slowly so that the powder tumbles gently off the walls to ensure good mixing andthe solvent is evaporated.
* the flask is lowered into a water bath at 50-55 degree celcius when a good vaccum is developed
* an aliquot of 5 ml of lipid solution is introduced into the flask via the solvent inlet tube.
* the solvent is absorbed completely by the powder and the temperature of the bed is monitored.
* an evaporation process ,the temperature will decrease
* a second aliquot is introuced slowly when the temperature begins to rise again
* the temperature is allowed to rise to 30 degree celcius ,the vacuum is released and the dryning process is completed by connecting the flask containing the powder to lyophilizeer and leaving it evacuated overnight at room temperature
*the powder is transfered into a 10 ml glass vial containing 600mg solid each (100 mg lipid and 500mg sorbital support ) flused with nitrogen and sealed well and stored.
Sonication:
*at high energy level ,the average size of the vesicles is further redused
* this was first achieved onexposer of MLVS fultrasonic irradiation and still remains the method most widely used for producing small vesicels
*there are two methods of sonication based on the use of either probe or bath ultrasonic disintegrators .
*the probe is employed for dispersions. Which require higher energy in a small volume while the bath is more suitable for large volume of diluted lipids.
French pressure cell liposome :
*The French press [42] originally was established for breaking up cells under milder and more appropriate conditions compared to the ultrasound techniques, because lipids as well as proteins or other sensitive compounds might be degraded during the sonication procedure.
* This system is normally used in the volume of 1 to 40 mL and therefore is not suitable for large-scale production.
*However, a scale-up-based strategy on this technique was established as the microfluidization. This continuous and scaleable variation of the French press technique enforcesdownsizing of Liposomes by collision of larger vesicles at high pressure in the interaction chamber of the microfluidizer.
Membrane extrution:
*The most prominent scalable downsizing method is the extrusion. Size reduction is managed under mild and more reproducible conditions compared to those discussed above.
* In this method, preformed vesicles are forced through defined membranes by a much lower pressure as described in the French press method. Extrusion through polycarbonate filters was first published by Olson et al. in 1979 [45]. Mayer et al. [19]
* performed extensive studies on varying lipid compositions and the influence on extrusion behavior and membrane properties.
* Depending on the apparatus and scale, the diameters of these membranes range from 25 to 142 mm. Lipex Biomembranes Inc., now Northern Lipids Inc., invented a vessel system for extrusion which is marketed from the mL scale to several liters. As suggested for all downsizing methods, liposomes should be extruded above the Tc of the lipid composition; this system can be tempered. The Lipex extruder system is available in a jacketed mode to allow extrusion at higher temperatures.
Freez thaw method:
*the method is based upon freezing of a unilamellar dispersion and then thawing by standing at room temperature for 15 min and finally subjecting to brief sonication cycle .
*thus the process rupture and fuses suvs during which the solute equilibrates between inside and outside and the liposome themselves fuse and increase markedly in size.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Solvent dispersion method:
Ethanol injection:
*This method has been reported as one of the alternatives used for the preparation of suvs without sonication .
*an etenol solution of lipids is injected rapidely through a fine needle into an excess of saline or other aqueous medium .
*the rate of the injection is usally sufficient to achieve complete mixing ,so that the ethanol is diluted almost instantaneously in water and phospholipid molecule are dispersed evenly troughout the medium.
*this procedure yields a high proportion of suvs
* this method is extremely simple and has low risk of degradation ofsensitive lipids.
Ether enjection:
*ether injection method is similar to the ethanol injection in many respects.
*it involves injecting the immiscible organic solution very slowly into an aqueous phase through a narrow needle at the temperature of vapourizing the organic solvent
*this method may also treat sensitive lipids very gently
*it has little risk of causing oxidative degradation provided ether is free from peroxides.
Reverse phare evaporation method:
*Similarly to the above presented injection methods, lipid is hydrated via solubilization in an organic phase followed by introduction into an aqueous phase.
*The organic phase should be immiscible with the aqueous phase, thus an oil/water emulsion is created, which is diluted with further aqueous phase for liposome formation
* The advantage of this very popular preparation technique is a very high encapsulation rate up to 50%. One variation of the microemulsion technique, the double emulsion technique, further improves the encapsulation rates and results in unilamellar liposomes .
* A possible drawback of this efficient method is the remaining solvent or the proof of their absence especially for using them for pharmaceutical purposes.
* The other important issue is large-scale production which might be feasible if appropriate shear mixing devices for the creation of the microemulsion and pumps for the dilution step are available.
Double emulsion methods:
This method requires two steps for the preparationof liposomes,
*first the inner leaflet of the bilayer then the outer half .
*the common features of this method is the formation of” water In oil” emulsion by introduction of a small quantity of aqueous medium containing materisl to be entrapped into large volume of immisible organic solution of lipid .this was followed by mechanical agitation to brek up the aqueous phase into microscopic water droplets
*these droplets are stabilized by the presense of phospholipid monolayer at the interface.
*the size of droplet is determined by the intensity of mechanical energy used to form the emulsion and amount of lipid relative to the volume of aqueous phase since each droplet requires a complete monolayer of phospholipid covering its surface in order to prevent the possible coalescence with other droplets(59,60)
Application:
Applications of liposomes in the sciences.
Discipline Application
* Mathematics :Topology of two-dimensional surfaces in three-dimensional space governed only by bilayer elasticity
* Physics: Aggregation behaviour, fractals, soft and high-strength materials
* Biophysics: Permeability, phase transitions in two-dimensions, photophysics
* Physical Chemistry: Colloid behaviour in a system of well-defined physical characteristics, inter and intra-aggregate forces, DLVO
* Chemistry: Photochemistry, artificial photosynthesis, catalysis,
* Biochemistry: Reconstitution of membrane proteins into artificial membranes
* Biology: Model biological membranes, cell function, fusion, recognition
* Pharmaceutics: Studies of drug action
* Medicine: Drug-delivery and medical diagnostics, gene therapy(61).
1.Conventional liposomes:-
For historical reasons we shallconventional liposomes distinguish between conventional liposomes and liposomes with altered surface properties.
ØThe first generation of liposomes includes various lipid compositions which changed the physicochemical properties of liposomes in a variety of different ways, but could not significantly alter their biological properties upon intravenous administration which is the most widely used route in medical applications.
ØTherefore, the optimistic goals of antibody sensitised liposomes (immunoliposomes as ‘guided missiles’), which gave often very encouraging results in in vitro studies – which are in general performed in the absence of immunoglobulins, complement components, and macrophages – failed in in vivo applications.
ØThe first condition for the immunoliposome concept to work is therefore that the escape the clearance by the mononuclear phagocytic system.
ØThis was made possible by the introduction of sterically stabilized liposomes in which the presence of surface grafted hydrophilic polymers substantially prolongs the liposome blood circulation times, probably due to reduced interactions with the components of the immune system.
This reduction arises from the presence of a steric barrier which prevents adsorption or hydrophobic binding of immune system components onto the foreign body. The liposomes with altered surfaces therefore include sterically stabilized liposomes and immunoliposomes.
ØWith respect to sterically stabilized immunoliposomes one should add a note of caution. Even liposomes with prolonged circulation in blood are not likely to be as widely applicable as many researchers envision(ed)(62).
2.Liposomes in parasitic diseases and infections:-
ØSince conventional liposomes are digested by phagocytic cells in the body after intravenous administration, they are ideal vehicles for the targetting of drug molecules into these macrophages.
ØThe best known examples of this ‘Trojan horse-like’ mechanism are several parasitic diseases which normally reside in the cell of mononuclear phagocytic system. They include leishmaniasis and several fungal infections. Leishmaniasis is a parasitic infection of macrophages which affects over 100 million people in tropical regions and is often fatal.
ØThe efficacious dose of drugs, mostly different antimonials, is not much lower than the toxic one. Liposomes accumulate in the very same cell population which is infected and therefore offer an ideal drug delivery vehicle (63). Indeed, the therapeutic index was increased in rodents as much as several hundred times upon administration of the drug in various liposomes.
ØThe best results reported so far in human therapy are probably liposomes as carriers for Amphotericin B in antifungal therapies.
ØThis drug is the drug of choice in disseminated fungal infections which often parallel compromised immune system, chemotherapy, or AIDS and are frequently fatal. Unfortunately, the drug itself is very toxic and its dosage is limited due to its nephro- and neuro-toxicity.
ØThese toxicities are normally correlated with the size of the drug molecule or its complex and obviously liposome encapsulation prevents accumulation of drug in these organs and drastically reduces toxicity (63).
ØSimilar approaches can be implemented in antibacterial, and antiviral therapy (64). Most of the antibiotics, however, are orally available and liposome encapsulation can be considered only in the case of very potent and toxic ones which are administered parenterally.
ØThe preparation of antibiotics loaded liposomes at reasonably high drug to lipid ratios may not be easy because of the interactions of these molecules with bilayers and high densities of their aqueous solutions which often force liposomes to float as a creamy layer on the top of the tube. Several other routes, such as topical or pulmonary (by inhalation) are being considered also.
ØLiposome encapsulated antivirals such as acyclovir, ribavarin, or azide thymidine (AZT) have also shown reduced toxicity and currently more detailed experiments are being performed with respect to their efficacy(65).
3.Macrophage activation and vaccination:-
ØSome natural toxins induce strong macrophage response which results in macrophage activation. This can be duplicated and improved by the use of liposomes because small molecules with immunogenic properties (haptens) cannot induce immune response without being attached to a larger particle.
ØFor instance, liposomes containing muramyl tripeptide, the smallest bacterial cell wall subunit with immunogenic properties cause macrophage activation. Activated macrophages are larger and contain more granulomae and lysosome material. Their state lasts for a few days during which they show enhanced tumouricidal, virocidal, and microbicidal activity.
ØEarly expectations in antitumour activity turned out to be too optimistic due to the simple fact that the number of free circulating macrophages is too small for an effective therapy. In cancer therapy, however, surgery or radiotherapy often do(es) not remove all the tumour cells and in these cases, when tumour burden is low, this therapy is very promising for complete eradication of malignant cells. Activation of macrophages was proven useful in the treatment of viral, bacterial, and fungal infections as well. Synergy between encapsulated immunomodulators and other activating factors such as cytokines and lymphokines, including interferon, (66).
ØMacrophages are involved also in the process of immunisation. Many molecules, however, do not induce an immune response because they are too small. In order to do so, they must be attached to larger particles. Normally this is done by administration of alum or killed bacteria and obviously liposomes offer an elegant alternative (67). Indeed, liposomes are used in animal vaccination already since 1988, while human vaccinations against malaria are now in clinical trials (68).
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
4. Liposomes in anticancer therapy:-
ØMany different liposome formulations of various anticancer agents were shown to be less toxic than the free drug( 69).
ØAnthracyclines are drugs which stop the growth of dividing cells by intercalating into the DNA and therefore kill predominantly quickly dividing cells. These cells are in tumours, but also in gastrointestinal mucosa, hair, and blood cells and therefore this class of drugs is very toxic.
ØThe most used and studied is Adriamycin (commercial name for Doxorubicin HCl). In addition to the above mentioned acute toxicities its dosage is limited by its cumulative cardiotoxicity. Many different formulations were tried. In most cases the toxicity was reduced about 50%. This includes both, short term and chronic toxicities because liposome encapsulation reduces the distribution of the drug molecules towards those tissues.
Ø For the same reason, on the other hand, the efficacy was in many cases compromised due to the reduced bioavailability of the drug, especially if the tumour was not phagocytic, or located in the organs of mononuclear phagocytic system. In some cases, such as systemic lymphoma, the effect of liposome encapsulation showed enhanced efficacy due to the sustained release effect, i.e. longer presence of therapeutic concentrations in the circulation (70) while in several other cases the sequestration of the drug into tissues of mononuclear phagocytic system actually reduced its efficacy. Applications in man showed in general reduced toxicity, better tolerability of administration with not too encouraging efficacy. Several different formulations are in different phases of clinical studies and show mixed results (71).
5.Liposomal formulations for clinical application:-
According to Crommelin and Storm212, the following quality-control assays should be applied to liposomal formulations:
Basic characterization assays:pH; osmolarity; trapped volume; phospholipid concentration; phospholipid composition; phospholipid acyl chain composition; cholesterol concentration; active compound concentration; residual organic solvents and heavy metals; active compound/phospholipid ratio; proton or ion gradient before and after remote loading.
Chemical stability assays:phospholipid hydrolysis; non-esterified fatty acid concentration; phospholipid acyl chain auto-oxidation; cholesterol autoxidation; active compound degradation.
Physical characterization assays:appearance; vesicle size distribution; sub-micron range;micron range; electrical surface potential and surface pH; zetapotential; thermotropic behaviour,phase transition, and phase separation; percentage of free drug(72).
6.Magnetic liposomes.: -
An interesting approach for targeted drug delivery under the action of magnetic field is the use of liposomes loaded with a drug and a ferromagnetic material. In one example, magnetic liposomes containing doxorubicin were intravenously administered to osteosarcoma-bearing hamsters. When the tumour-implanted limb was placed between two poles of a 0.4 Tesla magnet, the application of the field for 60 minutes resulted in a fourfold increase in drug concentration in the tumour(73). In the same osteosarcoma model in which the magnet was implanted into the tumour, magnetic liposomes loaded with adriamycin demonstrated better accumulation in tumour vasculature and resulted in enhanced tumour-growth inhibition)(74). Intravenous injection in rats of liposomes loaded with 99mTc-albumin and magnetite resulted in a 25-fold increase in accumulated radioactivity in the right kidney, near which a SmComagnet was implanted, compared with the control left kidney(75). This might become a promising way of drug targeting by liposomes.
7.Liposomes in diagnostic imaging:-
The use of liposomes for the delivery of imaging agents for all imaging modalities has a long history(76).
The relative efficacy of entrapment of contrast materials into different liposomes, as well as the advantages and disadvantages of various liposome types, have been discussed by Tilcock(77).
Liposomal contrast agents have been used for experimental diagnostic imaging of liver, spleen, brain, cardio-vascular system, tumours, inflammation and infections151,153.GAMMA-SCINTIGRAPHY and MRI both require a sufficient quantity of radionuclide or paramagnetic metal to be associated with the liposome.
There are two possible routes to improve the efficacy of liposomes as contrast mediums for gamma-scintigraphy and MRI:
A.increasing the quantity of carrier-associated reporter metal (such as 111In or Gd), and/or enhancing the signal intensity.
B.To increase the load of liposomes with reporter metals, amphiphilic chelating polymers, such as N,α-(DTPA polylysyl)glutaryl phosphatidyl ethanolamine, were introduced(78).
These polymers easily incorporate into the liposomal membrane and markedly increase the number of chelated Gd or In atoms attached to a single lipid anchor. In the case of MRI, metal atoms chelated into these groups are directly exposed to the water environment, which enhances the signal intensity of the paramagnetic ions and leads to corresponding enhancement of the vesicle contrast properties.
The overall performance of chelating polymer-bearing liposomes might be further improved by additional incorporation of amphiphilic PEG into the liposome membrane, because of the presence of the increased concentration of PEG-associated water protons in the close vicinity of chelated Gd ions located on the liposomal membrane.
In addition to the enhanced RELAXIVITY, the coating of liposome surface with PEG polymer can help in preventing the contrast agent being taken up at the site of injection by resident phagocytic cells. This approach results in efficient liposomal contrast agents for MRI of the blood pool(79) .
MRI using pH-responsive contrast liposomes allows for the visualization of pathological areas with decreased pH values(80). Liposomes loaded with contrast agent have also used for the in vivo monitoring of tissue pharmacokinetics of liposomal drugs in mice(81).
8.ATP liposomes.:-
There is interest in liposomal forms of ‘bioenergic’ substrates, such as ATP, and some encouraging results with ATP-loaded liposomes in various in vitro and in vivo models have been reported.ATP liposomes were shown to protect human endothelial cells from energy failure in a cell culture model of sepsis191. In a brain ischaemia model, the use of the liposomal ATP increased the number of ischaemic episodes tolerated before brain electrical silence and death192. In a HYPOVOLEMIC shock-reperfusion model in rats, the administration of ATP liposomes provided effective protection to the liver(82).
ATP liposomes also improved the rat liver energy state and metabolism during the cold storage preservation(83). Similar properties were also demonstrated for the liposomal coenzyme Q10 . Interestingly, biodistribution studies with the ATP liposomes demonstrated significant accumulation in the damaged myocardium196. Recently,ATP-loaded liposomes were shown to effectively preserve mechanical properties of the heart under ischaemic conditions in an isolated rat heart model(84).
ATP-loaded immunoliposomes have also been prepared that possess specific affinity towards myosin — that is, which are capable of specifically recognizing hypoxic cells(85).
9.Liposomes in photo-dynamic therapy:-
Photo-dynamic therapy (PDT) is a rapidly developing modality for the treatment of superficial tumours, in which photosensitizing agents are used for the photo-chemical eradication of malignant cells.
In PDT, liposomes are used both as drug carriers and enhancers, and a review on the use of liposomes in PDT has recently been published(86). Targeting as well as the controlled release of photosensitizing agent in tumours might still further enhance the outcome of the liposome-mediated PDT.
A benzoporphyrin derivative encapsulated in polycation liposomes modified with cetyl-polyethyleneimine was used for antiangiogenic PDT.This drug, encapsulated in such liposomes, was better internalized by human umbilical vein endothelial cells and was found in the intranuclear region and associated with mitochondria(87).
The commercial liposomal preparation of the benzoporphyrin derivative monoacid ring A (Visudyne; Novartis) is active against tumours in sarcoma-bearing mice(88).PDT with liposomal photofrin provides better results against human gastric cancer in mice than is achieved with free drug(89). Another porphyrin derivative (SIM01) in dimyristoylphosphatidylcholine iposomes also produces better results in PDT, mainly due to better accumulation in the tumour (human adenocarcinoma in nude mice)(90). Liposomal meso-tetrakis-phenylporphyrin is effective in PDT of human amelanotic melanoma in nude mice(91).The interest in this area of liposomology is still growing.
10.Liposomes modified with cell-penetrating peptides:-
. A new approach to drug delivery has recently emerged, which is based on the use of certain viral proteins that have the ability to penetrate into cells (the so-called ‘protein transduction’phenomenon). The transactivating transcriptional activator (TAT) protein from HIV-1 enters various cells when added to the surrounding media(92).
Recent data indicate that there is more than one mechanism used by cell-penetrating peptides and proteins (CPP) and CPP-mediated intracellular delivery of various molecules and particles. TAT-mediated intracellular delivery of large molecules and nanoparticles occurs through energy-dependent macropinocytosis, with subsequent enhanced escape from endosome into the cell cytoplasm(93), whereas individual CPPs or CPPconjugated small molecules penetrate cells via electrostatic interactions and hydrogen bonding and and the penetration does not seem to be associated with metabolic energy (that is, it is a purely physical, not biological, process)(94). Traversing cellular membranes represents amajor barrier for the efficient delivery of macromolecules into cells, and therefore CPPs, whatever their mechanism of action, could serve to transport various drugs and even drug-loaded pharmaceutical carriers into mammalian cells in vitro and in vivo. It has been demonstrated that relatively large particles, such as liposomes, can be delivered into various cells by several TAT-peptide or other CPP molecules attached to the liposome surface(94,95). Complexes of TAT-peptide liposomes with a plasmid (plasmid pEGFP-N1, which encodes the green fluorescent protein) were used for successful in vitro transfection of various tumour and normal cells, as well as for in vivo transfection of tumour cells in mice bearing Lewis lung carcinoma(96). Whatever mechanisms underlie the TAT-mediated delivery of large cargo such as liposomes into cells, the covalent coupling of TAT-peptides to microparticulate drug carriers could provide an efficient tool for the cytosolic delivery of various drugs and DNA in vitro and even in vivo in certain protocols of local treatment.
CONCLUSION:
liposome are now used to deliver certain vaccines,enzymes and drugs to the body .when used in the delivery of certain cancer drugs,liposomes help to shield healthy cells from the drugs toxicity and prevent their concentration in vulnerable tissues(e.g kidney ,liver ),lessening or elimination the common side effects of nausea,fatigue and hair lose.
Liposome are especially effective in treating diseases that efferts phagocytes .also used to carry genes into cells and can be administered by various route.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
REFRENCES:
1.target and controlled drug delivery –novel carrier systems by S.P Vyas and S.K Khar.
Controlled and novel drug delivery systems,chapter 15-liposome as a drug carriers by Sanjay.k.jain and N.K.Jain(2-12).
13. Lichtenberg,D., and Barenholz, Y., 1988. In: Glick, D. (Ed.) Methods of biological analysis, Vol. 33, John Wiley and Sons Inc., New York, pp. 337-461.
14. Nassander, U.K., Storm, G., Peeters, P.A.M., Crommelin, D.J.A., 1990. Liposomes. In: Chosin, M. and Langer, R., (Eds.), Biodegradeable Polymers as Drug Delivery Systems, Marcel Dekker Inc., New York, pp. 261-338.
15. Lasic, D.D., 1998. Novel application of liposomes. Tibitech. 16 307-321.
16. Gregoriadis,G., 1988. In: Gregoriadis, G., (Ed.), Liposomes as drug carriers: recent trends and progress, Chicester, John Wiley, pp. 52-61.
17. Szoka,F.C., 1991, In: Wilschut, L., Hockstra, D., (Eds), Membrane Fusion, Marcel Dekker Inc., New York, pp. 845-890.
18. Chrai,S.S. Murari, R., Ahmad, I., 2001. Liposomes (a review) part one: manufacturing issues. Bio. Pharm. 11, 10-14.
19. Lasic,D.D., 1993. In: Liposomes: From Biophysics to applications, Amsterdam, Elsevier, NY, pp. 9-11.
20. Lipowsky, R., 1991. The confirmation of membrane. Nature 349, 475-481. IJCPR August-October 2010; 1(2) 15 Sharma Vijay et.al./ Liposomes: Present Prospective and Future Challenge.
21. Hope, M.J., Kitson, C,N., 1993. Liposomes. A perspective for dermatologists. Dermatol. Clin. 11, 143-154.
22. Eibl, H., 1980. Synthesis of glycerophospholipids, Chem. Phys. Lipids 26, 405-429.
23. Weiner, N., Martin, F., Riaz, M., 1989. Lipoomes as drug delivery system. Drug Develop. Ind. Pharm. 15, 1523-1554.
24. Szoke, F. Jr., Papahadjopoulos, D., 1980. Comparative properties and methods of preparation of lipid vesicles (liposomes). Ann. Rev. Biophys. Bioeng. 9, 467-508.
25. New, R.R.C., 1990. Preparation of liposomes. In: New R.R.C., (Ed.), Lipsomes: A Practical Approach, IRL Oxford University Press, New York, pp. 33-104.
26. Mezei, M., 1993. Liposomes and the skin. In: Gregoriadis, G., Florence, A.T., Patel, H.M., (Eds.), Liposomes in Drug Delivery, Harwood Academic Publishers pp. 125-135.
27. Mayer, L.D., Hope, M.J., Cullis, P.R., Janoff, A.S., 1985. Solute distributions and trapping efficiencies observed in freeze-thawed multilmellar vesicles. Biochim. Biophys. Acta 817, 193-196.
28. Vyas, S.P., Khar, R.K. 2006. Targeted And Controlled Drug Delivery: Novel Carrier Systems. Edition 1, CBS Publishers & Distributor, New Delhi.pp.421-427.
IJCPR
29. Bangham, A.D. and R.W. Horne, 1964, Negative staining of phospholipids and their structured modofication by surface active agents as observed in the electron microscope, J. Mol. Biol. 8, 660–668.
30. Papahadjopoulos, D. (ed.), 1978, Liposomes and their use in biology and medicine, Ann. NY Acad. Sci. 308, 1–412.
31. Lasic, D.D., 1992, Liposomes, Am. Sci. 80, 20–31.
32. Lipowsky, R., 1992, The conformation of membranes, Nature 349, 475–481.
33. Lasic, D.D., 1993, Liposomes: From Physics to Applications (Elsevier, Amsterdam) 1993.
34 see articles by E. Sackmann, R. Lipowsky, V.A. Parsegian, E. Evans and Helfrich, in this volume.
35. Svetina, S. and B. Zeks, 1982, Bilayer couple as a possible mechanism of biological shape formation, Biomed. Biophys. Acta 44, 979–986.
36. Berndt, K., J. Kaes, R. Lipowsky, E. Sackmann and U. Seifert, 1990, Shape transformations of giant vesicles: extreme sensitivity to bilayer asymmetry, Europhys. Lett. 13, 659–664.
37. Kaes, J. and E. Sackmann, 1991, Shape transitions and shape stability of giant phospholipid vesicles in pure water induced by area-to-volume changes, Biophys. J. 60, 825–844.
38. Bloom, M., O. Mouritsen and E. Evans, 1991, Physical properties of the fluid lipid bilayer component of cell membranes: A perspective, Q. Rev. Biophys. 24, 293–397.
39. Helfrich, W., 1974, The size of sonicated vesicles, Phys. Lett. 50a, 115–116. 12. Lasic, D.D., 1988, The mechanism of liposome formation. A review, Biochem. J. 256, 1–11.
40. Lasic, D.D., 1991, Formation of membranes, Nature 351, 163.
41. Fendler, J.H., 1987, Atomic and molecular clusters in membrane mimetic chemistry, Chem. Rev. 87, 877–899.
42. Cornelius, F., 1991, Functional reconstitution of the sodium pump. Kinetics and exchange reactions performed by reconstituted Na/K ATPase, Biochim. Biophys. Acta 1071, 19–66.
43. Villalobo, A., 1991, Reconstitution of ion-motive transport ATPases in artificial lipid membranes, Biochim. Biophys. Acta 1071, 1–48. Applications of liposomes 517
44. Lasic, D.D., P.M. Frederik, M.C.A. Stuart, Y. Barenholz and T.J. McIntosh, 1992, Gelation of liposome interior. A novel method for drug encapsulation, FEBS Lett. 312, 255–258.
45. Nassander, U.K., P.A. Steerenberg, P.A. Poppe, G. Storm, L.G. Poeis, W.H. de Jong and D.J.A. Crommelin, In vivo targetting of OV-TL3 immunoliposomes to ascitic ovarian carcinoma cells (OVCAR- 3) in athymic nude mice, Cancer Res. 52, 646–653.
46. New, R.R.C., S.M. Chance, S.C. Thomas and W. Peters, 1978, Nature antileshmanial activity of antimonials entrapped in liposomes, 272, 55–58.
47. Lopez-Berestein, G., V. Fainstein, R. Hopter, K.R. Mehta, M. Sullivan, M. Keating, M. Luna and E.M. Hersh, 1985, Liposomal Amphotericin B for the treatment of systemic fungal infections in patients with cancer, J. Infect. Diseases 151, 704–710.
48. Lasic, D.D., 1992, Mixed micelles in drug delivery, Nature 355, 279–280. 22. Svenson, C.E., M.C. Popescu and R.C. Ginsberg, 1988, Liposome treatments of viral, bact and protozoal infections, Crit. Rev. Microbiol. 15, S1–31.
49. Fidler, I.J., D. Fan and Y. Ichinose, 1989, Potent in situ activation of murine lung macrophages and therapy of melanoma metastases, Invas. Metastasis 9, 75–88.
50. Gregoriadis, G., 1990, Immunological adjuvants: A role for liposomes, Immunol. Today 11, 89–97.
51. Alving, C.R., Liposomes as carriers of antigens and adjuvants, J. Immunol. Methods, in press.
52. Gabizon, A., 1989, Liposomes as a drug delivery system in cancer therapy, in: Drug Carrier Systems, eds F.H.D. Roerdink and A.M. Kron (Wiley, Chichester) pp. 185–211.
53. Storm, G., F.H. Roerdink, P.A. Steerenberg, W.H. de Jong and D.J.A. Crommelin, 1987, Influence of lipid composition on the antitumor activity exerted by doxorubicin containing liposomes in a rat solid tumor model, Cancer Res. 47, 3366–3372.
54. Lopez-Berestein, G. and I.J. Fidler (eds), 1989, Liposomes in the Treatment of Infectious Diseases and Cancer (A. Liss, New York).
55. Williams, B.D., M.M. Sullivan, K.E. Williams, J.R. Williams and J.R. Morgan, 1986, Imaging in rheumatoid arthritis using liposomes labeled with technetium, Br. Med. J. 293, 1144–1145.
56. McCalden, T., 1990, Particulate systems for drug delivery to the lung, Adv. Drug Del. Rev. 5, 253–263.
57. Fielding, R.M. and R.M. Abra, 1992, Factors affecting the release rate of terbutaline from liposome formulations after intratracheal instillation in the guinea pig model, Pharm. Res. 9, 220–222.
58. Hamori, C.J., D.D. Lasic, H.J. Vreman and D.K. Stevenson, 1993, Targeting zinc protoporphyrin liposomes to the spleen using reticuloendothelial blockade with blank liposomes, Pediatric Research,
59. Allen, T.M. and A.Chonn, 1986, Large unilamellar liposomes with low uptake by the reticuloendothelial system, FEBS Lett. 223, 42–46.
60. Gabizon, A. and D. Papahadjopoulos, 1988, Liposome formulations with prolonged circulation times in blood and enhanced uptake by tumors, Proc. Nat. Acad. Sci. USA 85, 6949–6953.
61. Woodle, M.C. and D.D. Lasic, 1992, Sterically stabilized vesicles, Biochim. Biophys. Acta 1113, 171–199.
62. Lasic, D.D., F.J. Martin, A. Gabizon, K.S. Huang and D. Papahadjopoulos, 1991, Sterically stabilized vesicles: A hypothesis on the molecular origin of extended circulation times, Biochim. Biophys. Acta 1070, 187–192.
63. de Gennes, P.G., 1987, Polymers at an interface: A simplified view, Adv. Colloid Interface Sci. 27, 189–209.
64. Needham, D., T.J. MacIntosh and D.D. Lasic, 1992, Repulsive interactions and mechanical stability of polymer-grafted lipid membranes, Biochim. Biophys. Acta 1108, 40–48.
65. Needham, D., K. Hristova, T.J. McIntosh, M. Dewhirst, N. Wu and D.D. Lasic, 1992, Polymergrafted liposomes: Physical basis for the stealth property, J. Liposome Res. 2, 411–430.
66. de Gennes, P.G., 1991, A second type of phase separation in polymer solutions, C.R. Acad. Sci., 313-II, 1117–1122.
67. de Gennes, P.G., 1992, Private communication.
68. Lasic, D.D. and D. Needham, Stealth liposomes: A prototypic biomaterial, Chem. Rev., in press.
69. Gabizon, A., 1992, Selective tumor localization and improved therapeutic efficacy of anthracyclines encapsulated in long circulating liposomes, Cancer Res. 52, 891–896. 518 D.D. Lasic
70. Papahadjopoulos, D., T.M. Allen, A. Gabizon, E. Mayhew, S.K. Huang, M.C. Woodle, D.D. Lasic, C. Redemann and F.J. Martin, 1991, Sterically stabilized vesicles: Improvements in pharmacokinetics and antitumor therapeutic efficacy, Proc. Nat. Acad. Sci. USA 88, 11460–11464.
71. Mayhew, E.G., D.D. Lasic, S. Babbar and F.J. Martin, 1992, Pharmacokinetics and antitumor activity of epirubicin encapsulated in long circulating liposomes incorporating a polyethylene glycol derivatized phospholipid, Int. J. Cancer 51, 302–309.
72. J. Vaage, E. Mayhew, D.D. Lasic and F.J. Martin, 1992, Therapy of primary and metastatic mouse mammary carcinoma with doxorubicin encapsulated in long circulating liposomes, Int. J. Cancer, 51, 942–948.
73. Huang, S.K., E. Mayhew, S. Gilani, D.D. Lasic, F.J. Martin and D. Papahadjopoulos, 1992, Pharmacokinetics and therapeutics of sterically stabilized liposomes in mice bearing C-26 colon carcinoma, Cancer Res. 52, 6774–6781.
74. Allen, T.M., T. Mehra, C. Hansen and Y. Chin, 1992, Stealth liposome: An improved sustained release system for arabinofuranosylcytosine, Cancer. Res. 52, 2431–2439.
75. Bakker-Woudenberg, I., A.F. Lokersee, M. ten Kate and G. Storm, 1992, Enhanced localization of liposomes with prolonged blood circulation times in infected lung tissue, Biochim. Biophys. Acta, 1138, 318.
76. Woodle, M.C., G. Storm, M.S. Newman, J.J. Jekott, L.R. Collins, F.J. Martin and F.C. Szoka, 1992, Prolonged systemic delivery of peptide drugs by long circulating liposomes, Pharm. Res. 9, 260–265.
77. Martin, F., A. Gabizon and F.J. Martin (Eds), 1992, Human pharmacokinetics of doxil (stealth liposome encapsulated doxorubicin), in: UCLA Symposium: Prevention and Treatment of AIDS, Book of Abstracts, Keystone, CO.
78. Bogner J.R., F.D. Goebel, 1995, Phase II open-label trial of doxil (stealth liposomal doxorubicin) in advanced AIDS-KS, in: VIII International Conference on AIDS, in ref. 51, in press. Boca Raton.
79. Northfelt, D.W., L. Kaplan, J. Russell, P.A. Volberding, F.J. Martin, Single dose pharmacokinetics, safety and tumor localization study of doxil in AIDS patients with Kaposi’s sarcoma, in: VIII International Conference on AIDS, in ref.
80. Nicolau, C. and A. Cudd, 1989, Liposomes as carriers of DNA, Crit. Rev. Therap. Drug Carr. Systems 6, 239–271.
81. Fraley, R.T, S. Subramani, P. Berg and D. Papahadjopoulos, 1980, Introduction of liposome encapsulated SV40DNA into cells, J. Biol. Chem. 255, 10431–10435.
82. Lurquin, P., 1979, Entrapment of plasmid DNA by liposomes and their interaction with plant protoplasts, Nucl. Acids Res. 6, 3773–3777.
83. Felgner, P., T.R. Gadek, M. Holm, R. Roman, M.Wenz, J.P. Northrop, G. Ringold andM. Danielsen, 1987, Lipofectin: A highly efficient, lipid mediated DNA transfection procedure, Proc. Nat. Acad. Sci. USA .
84. Rose, J.K., L. Buoncore and M.A. Whitt, 1991, A new cationic liposome reagent mediating nearly quantitative transfection of animal cells, Biotechniques 10, 520–525.
85. Gao, X. and L. Huang, 1991, A novel cationic liposome reagent for efficient transfection of mammalian cells, Biophys. Biochem. Res. Commun. 179, 280–285.
86. Stribling, R., E. Brunette, D. Liggett, K. Gaenslar and R. Debs, 1992, Aerosol gene delivery in vivo, Proc. Nat. Acad. Sci. USA 89, 11277–11281.
87. Law, B.A., J.S. King, 1991, Use of liposomes for proteinase addition to cheddar cheese, J. Diary Res. 52, 183–188.
88. Alkhalaf, W., J.C. Piard, M. el Soda, J.C. Gripon, M. Desmezeaud and L. Vassal, 1988, Liposomes as proteinases carriers for the accelerated ripening of St. Paulin type cheese, J. Food Sci. 53, 1674–1679.
89. Kirby, C., 1990, Delivery systems for enzymes, Chem. Br., Sept. 1990, 847–851.
90. Tahibi, A., J.D. Sakurai, R. Mathur and D.F.H. Wallach, 1991, Novasome vesicles in extended pesticide formulation, Proc. Symp. Contr. Rel. Bioact. Mat. 18, 231–232. Applications of liposomes 519
91.. Gatt, S., J.H. Bercovier and Y. Barenholz, 1991, Use of liposomes to combat oil spills and their potential application to bioreclamation, in: On Site Bioreclamation, eds R.E. Hinchee and R.F. Olfenbuttel (Butterworth, Stoneham) pp. 293–312.
92. Dutton, G., 1993 (Feb.), The promise of liposomes, Gen. Engin. News, 13, 6–9.
93. Boroske, E., M. Elwenspoek and W. Helfrich, 1981, Osmotic shrinkage of giant egg-lecithin vesicles, Biophys. J. 34, 95–109.
94. Kuhl, T., D. Leckband, D.D. Lasic, J. Israelachvili, 1994, Biophys. J. 66, 1479.
95. Tsuchida E: Stabilized hemoglobin vesicles. Artif Cells Blood Substitute lmmobil Biorechnol 1994, 221467-477.
96. Zheng S, Zheng Y, Beissinger R: Efficacy, physial properties and pharmacokinetics of sterically-stabilized liposomc-cncap sulated hemo@bii. Artif Cells Slood Subsritu~e mmobil Biorechnol 1994, 22:487-501.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






