
 About Authors:
About Authors:
NIKHIL SAXENA, DR.K.R.PARDASANI, DR.USHA CHAUHAN
MAULANA AZAD NATIONAL INSTITUTE OF TECHNOLOGY,
BHOPAL
ABSTRACT
The electro negativity of the drug directly resembles its chemical nature towards the receptor. In order to achieve the desired response, the effective binding of drug is necessary. Based on the Lock key theory, every drug having a specific structure so that it can bind to a specific receptor. But for designing more efficient a new chemical entity for receptor as compared to the main drug, there is a requirement of some structural changes in the drug (ligand) molecules under structure activity relationship concept.
Here an attempt has been made to develop Insilco approach to make some structural changes on the basis of point mutation (changes occurs at a particular point) in the antiasthmatic drug namely salbutamol.
The effect of this mutation on binding affinity of the molecule is studied using docking energy (energy involved in orientation of one molecule to a second are when bound to each other to form a stable complex) calculations and logp values. Here eight different structures of the drug found are more electronegative as compared to the main drug candidate. Mutation was made at C-3a position in place of hydrogen. These were analyzed for binding affinity with target receptor. The mutated molecules which have high logp values and less energy are selected for further adme properties studies. Thus after prediction all adme properties and toxicity studies of the mutated drugs, the best mutation was bromine in place of hydrogen at C-3a position.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1221
INTRODUCTION:
The use of in silico predictive models of absorption, distribution, metabolism and elimination (ADME) and physicochemical properties is a major aid in this exercise, as it enables virtual molecules to be assessed across a broad range of properties from initial library generation, through to candidate selection. Of course, no measurement, whether in silico, in vitro or in vivo, is perfect and the uncertainties in any data should be explicitly taken into account when basing conclusions on test results. In addition, in the early stages of drug discovery [1] when designing a library that is lead seeking or building compound structure–activity relationships, the quality of any set of molecules should also be balanced against the chemical diversity covered. Here, a scheme is presented for achieving these goals based on a suite of predictive ADME models, probabilistic scoring and multiobjective optimisation for library design. The use of this platform for applications in lead identification and optimisation is illustrated. [2]
There have been considerable advances in the last few years in both the quantity and the quality of in silico ADMET property predictions. Most ADMET properties are now computable, and the accuracy of some of the software predictions for physicochemical properties in particular is close to that of measured data. There is, however, universal agreement that more good experimental ADMET data are needed for use in in silico model development, for models are only as good as the data on which they are based. Many data remain confidential but it is to be hoped that, with projects such as the Vitic toxicity database, being developed by Lhasa Limited, pharmaceutical companies will be prepared to release data to an ‘honest broker’ on a confidential basis, so that better in silico models can be developed. Incorporation of calculated ADMET properties into drug discovery and development is a multi-factorial problem and really needs a multi-factorial solution. Some progress is being made in this direction and it is hoped that within the foreseeable future software will be available for this purpose. [3]
The in silico metabolism simulators possessed several distinguishing features imparted in part by the nature of knowledge rules (algorithms) encoded within them and in part by the integration of QSAR libraries and computational engines.[4]
The success of any drug will depend on how closely it achieves an ideal combination of potency, selectivity, pharmacokinetics and safety. The key to achieving this success efficiently is to consider the overall balance of molecular properties of compounds against the ideal profile for the therapeutic indication from the earliest stages of a drug discovery project. The use of in silico predictive models of absorption, distribution, metabolism and elimination (ADME) and physicochemical properties is a major aid in this exercise, as it enables virtual molecules to be assessed across a broad range of properties from initial library generation, through to candidate selection.[5] Of course, no measurement, whether in silico, in vitro or in vivo, is perfect and the uncertainties in any data should be explicitly taken into account when basing conclusions on test results. In addition, in the early stages of drug discovery, when designing a library that is lead seeking or building compound structure–activity relationships, the quality of any set of molecules should also be balanced against the chemical diversity covered. Here, a scheme is presented for achieving these goals based on a suite of predictive ADME models, probabilistic scoring and multiobjective optimisation for library design.The use of this platform for applications in lead identification and optimisation is illustrated. [6]
DESIGN AND METHODOLOGY:
Here an attempt to use the receptor dependent methodology of QSAR technique on the basis of doing point mutation at a particular ligand site by using a chemical structure drawing software CHEMSKETCH. Just shown in the figure:
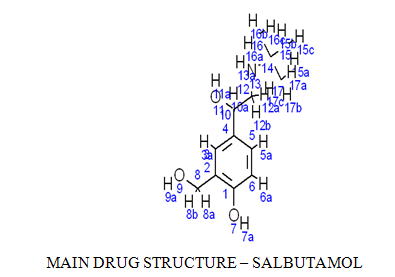
POINT MUTATION UNDER QSAR AT 3-aPOSITION HYDROGEN:
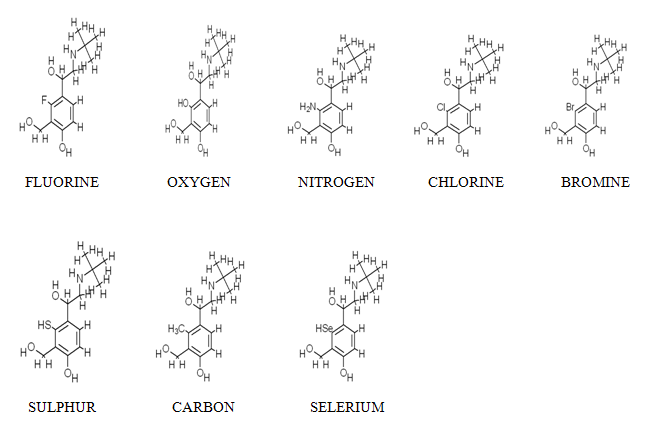
Now there are eight mutated drug structures are obtained by applying the SAR technology at the particular ligand site. After that the comparison among these mutated drugs will be involved in the docking operation between receptor (beta adrenergic receptor) and ligands (main drug candidate and modified drug candidates) for the energy calculation and the study about the ADME properties and Toxicities studies.
Thus on the basis of above study of the docking operation we can easily calculate the docking energy values in each and every combinations with various ligands and same target receptor. Now we will do comparative study of the result obtained by the docking operation and the selection of best suited drug candidate for the target.
[adsense:468x15:2204050025]
COMPARATIVE STUDY OF THE RESULTS
(TABLE NO.1)-FOR DOCKING ENERGIES AND LOGP VALUES
|
DRUG ( LIGAND) CANDIDATE |
DOCKING ENERGY (KJ/MOL) |
LOGP VALUES |
|
MAIN DRUG |
-23.49 |
0.01 |
|
POINT MUTATION IN DRUG (LIGAND) AT C-3a POSITION HYDROGEN |
DOCKING |
LOGP VALUES |
|
MUTATED DRUG-I (FLUORINE) MUTATED DRUG-II(OXYGEN) MUTATED DRUG-III(NITROGEN) MUTATED DRUG-IV(CHLORINE) MUTATED DRUG-V(BROMINE) MUTATED DRUG-VI(SULPHUR) MUTATED DRUG-VII(CARBON) MUTATED DRUG-VIII(SELERIUM) |
-25.70 -25.56 -26.67 -25.39 -25.39 -25.14 -26.67 -23.06 |
0.46 -0.71 -1.13 0.93 1.16 0.35 0.47 0.17 |
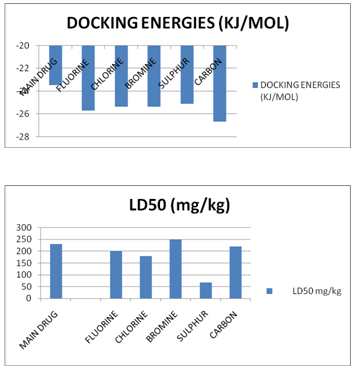
On the basis of above mentioned docking energies and logp values we can say that on doing the mutation c-3a position hydrogen into different more electronegative elements as compared to hydrogen we found five better mutated drugs as compared to the main drug. These are: Hydrogen to fluorine, chlorine, bromine, sulphur, and carbon. These five different mutated forms are involved to further study of the ADME properties for the selection of best suited drug
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
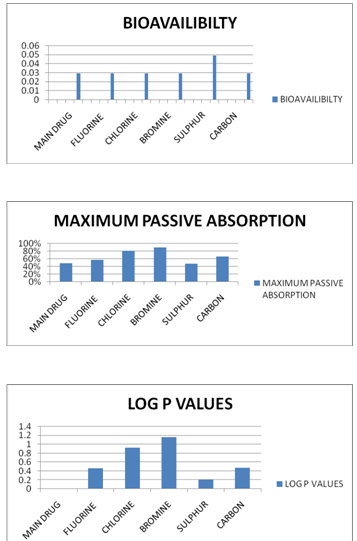
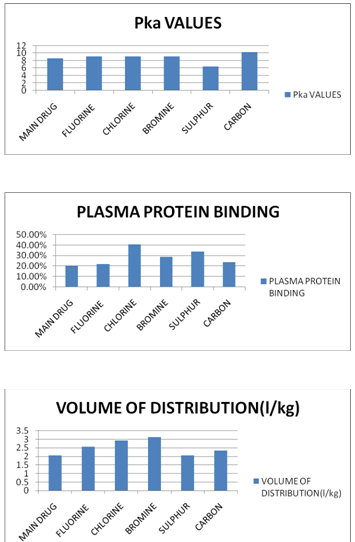
(TABLE NO.2) -FOR ADME PROPERTIES
|
DRUG CANDIDATE |
BIOAVAILABIITY |
MAXIMUM PASSIVE ABSORBTION |
LOG P VALUE |
Pka VALUE |
PLASMA PROTEIN BINDING |
VOLUME OF DISTRIBUTION (Vd) |
DOCKING ENERGY KJ/MOL |
|
MAIN DRUG |
%F (oral)>30%= 0.350 %F (oral)>70%= 0.029 |
49% |
0.01 |
St. acid=10.30 st base=8.60 |
20.09% |
2.07 l/kg |
-23.49 |
|
C-3a POSITION HYDROGEN TO FLUORINE |
%F (oral)>30%= 0.350 %F (oral)>70%= 0.029 |
58% |
0.46 |
9.10 |
22.07% |
2.56 l/kg |
-25.70 |
|
CHLORINE |
%F (oral)>30%= 0.350 %F (oral)>70%= 0.029 |
81% |
0.93 |
9.10 |
40.91% |
2.93 l/kg |
-25.39 |
|
BROMINE |
%F (oral)>30%= 0.350 %F (oral)>70%= 0.029 |
90% |
1.16 |
9.20 |
29.08% |
3.13 l/kg |
-25.39 |
|
SULPHUR |
%F (oral)>30%= 0.350 %F (oral)>70%= 0.049 |
47% |
0.20 |
6.40 |
34.11% |
2.05 l/kg |
-25.14 |
|
CARBON |
%F (oral)>30%= 0.350 %F (oral)>70%= 0.029 |
66% |
0.47 |
10.30 |
23.69% |
2.34 l/kg |
-26.67 |
(TABLE NO.3)-FOR TOXICITIES
|
DRUG |
LD50 |
PLD50 |
LOWER LIMIT |
UPPER LIMIT |
|
MAIN DRUG (SALBUTAMOL) |
230 mg/kg |
-0.05 |
-0.82 |
0.62 |
|
FLUORINE |
200 mg/kg |
0.11 |
-0.66 |
0.68 |
|
CHLORINE |
180 mg/kg |
0.19 |
-0.52 |
0.85 |
|
BROMINE |
250 mg/kg |
0.11 |
-0.66 |
0.75 |
|
SULPHUR |
67 mg/kg |
0.60 |
-0.12 |
1.40 |
|
CARBON |
220 mg/kg |
0.06 |
-0.79 |
0.68 |
Thus on the basis of above mentioned values of lethal dose we can easily conclude that the bromine mutated drug is best suited because the value of lethal dose is much high as compared to the main drug at a particular effective dose. The upper limit values are also high as compared to main drug candidate.
For the comparison of the toxicity studies we have to suppose the particular effective molecule and then compare with the lethal dose to calculate the therapeutic index for the selection of best suited drug candidate for the particular receptor. The drug which has the highest therapeutic index is the most suitable drug for the receptor at a particular effective dose of the drug in the human body.
AXIAL COMPARATIVE REPRESENTATION OF THE ADME PROPERTIES AND TOXICITIES
CONCLUSION:
It is very important to study about the drug designing methodology and you must have knowledge about the SAR (structure-activity-relationship) and what is the importance of SAR in the in-silico approach of drug designing. In the performed work we try to present a particular new pattern of drug designing. According to this new pattern or new method we develop a concept of point mutation under the study of QSAR methodologies. By means of this method we designed a new drug candidate which is more superior in each and every prospect as compared to the main drug candidate for the same receptor site.
These methods will have a growing impact of on drug design. In particular, the discovery of new lead structures and their optimization will profit by virtual screening.
REFERNCES:
1. A.R. Miedema “The electronegativity Parameter for Transition metals: Heat of Transformation and Charge Transfer in alloys,” journal of less common metals volume 32, issue I, July 1973, pg .117-136.
2. Andrew R. Leach, V.J.Gillet, “An introduction of Chemoinformatics”, pg 345-346, 1982.
3. Aristides Dolcometzidis, Lida Kalantzi, Nikoletta Fotaki, “ Predictive Model for Oral Absorption: Form in In-silico methods to integrated dynamic models”, Expert opinion on Drug metabolism and toxicity, volume III, No.4, Aug 2007, pg 491-505
4. Antonio Macchiarulo, Gabriele Costantino, Mirco Meniconi, Karin Pleban, Gerhard Ecker, Daniele Bellocchi, Robert Pellicciari, “ Insight into Phenylalanine Derivatives Recognition of VLA-4 Integrin: From a Pharmacophoric study to 3D-QSAR and Molecular modelling”, journal of medicinal chemistry, volume 24, 1986, pg . 46-48.
5. Barry A. Bunin, “Chemoinformatics: Theory Practice and Products”, pg 146-148, 1979.
6. B.L. Claus and D.J. Underwood, “Discovery Informatics: Its evolving role in drug discovery”, pg 957-966, 1978.
7. C.silipo, A Vittioria, “QSAR: Rational Approaches to the Design of Bioactive Compounds” pg 132-145, 1988.
8. Christel AS Bergstrom, “ Computational Models to Predict Aqueous Drug Solubility , Permeability and Intestinal Absorption, Expert opinion of drug metabolism and toxicology, volume 1, Dec 2005, No.4, pg 613-627.
9. David Livingstone, “Data Analysis for Chemist: Application to QSAR and Chemical Product Design”, pg 678-645, 1976.
10. David C.Yound , “ Computational Drug Design: A Guide for Computational and Medicinal Chemist,” pg 155-167, 1984.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








