

{ DOWNLOAD AS PDF }
ABOUT AUTHORS:
*Abhijit Chanda1, N.Ramalakshmi2, C.N Nalini2, S. Mahabubi1
1Department of Quality Assurance, Baxter (India) Pvt. Ltd
2Dept. of Pharmaceutical Analysis, C. L. Baid Metha College of Pharmacy
Chennai-600097, Tamilnadu, India
chandaabhijit91@gmail.com
ABSTRACT
Impurity profiling brings tremendous efforts in the group of analytical activities, the aim of which is the detection, identification/structure elucidation and quantitative determination of organic and inorganic impurities, as well as residual solvents in bulk drugs and pharmaceutical formulations. The control of impurities is currently a critical issue to the healthcare manufacturing. Various regulatory authorities like ICH, USFDA, UK-MHRA, CDSCO are emphasizing on the requirements and the identification of impurities in Active Pharmaceutical Ingredient’s (API’s) and as well as finished products. International Conference on Harmonization (ICH) formulated guidelines concerning the control and limit of impurities. To isolate and characterize impurities in pharmaceuticals diverse methods are used such as, capillary electrophoresis, gas–liquid chromatography, high performance liquid chromatography, solid-phase extraction methods, Ultraviolet Spectrometry, infrared spectroscopy, supercritical fluid extraction chromatography, mass spectroscopy, Nuclear magnetic resonance (NMR) spectroscopy etc. On the beginning of hyphenated techniques, the most browbeaten techniques for impurity profiling are Liquid Chromatography (LC)-Mass Spectroscopy (MS), LCNMR, LC-NMR-MS, GC-MS and fully automated Comprehensive Orthogonal Method Evaluation Technology (COMET). That is why it has plentiful claim in the field of drug design, monitoring quality, stability and as well as safety of the product.
INTRODUCTION
Over the last few decades there was much interest was compensated towards the quality of pharmaceuticals that enter into the market. The source of active pharmaceutical ingredient (APIs) of specific quality of the bulk drug industry which forms the base of all formulation based pharmaceuticals. So it is obligatory to carry outdynamic quality control checks in order to uphold the quality and purity of output from each industry. Impurity of pharmaceuticals may produce at any stage; it may occurduring synthesis, storage, due to side reaction, degradation, changes of any physiochemical property upon storage. So stability study of pharmaceutical also play key role in the field of impurity profiling.Impurities present more than 0.1% should be identified and quantified by selective methods.
Hence it is essential to know the structure of these impurities in the bulk drug in order to modify the reaction condition and to bring the quantity of impurity to an standard level[1,2] . Isolation, identification and quantification of impurities help us in various ways, to obtain a pure substance with less toxicity and, safety in drug therapy. Quantitative determination of these impurities could be used as a method for the quality control and validation of drug substances. Regulatory authorities and board such asICH (International Conference on Harmonization), USFDA (United sates Foods and Drug Administration), UK-MHRA (UK Medicines and Healthcare Products), Indian CDSCO (Central Drugs Standard Control Organization) Australian TGA (Therapeutic Goods Administration) etc.be firm on the impurity profiling of drugs. So it is very important to do qualification of impurities present in pharmaceuticals. Impurities in new drug substances are addressed from two perspectives:
Chemistry Aspects include classification and identification of impurities, report generation, listing of impurities in specifications, and a brief discussion of analytical procedures; and
Safety Aspects include specific guidance for qualifying those impurities that were not present, or were present at substantially lower levels, in batches of a new drug substance used in safety and clinical studies
Terminology for Impurity
Impurities have been named differently by a choice of scientists who deals with them. Terms that are used by official bodies such as compendia or that have been found acceptable by ICH and various regulatory bodies.
Impurity is defined by ICH as any component of the new drug substance which is not the chemical entity defined as the new drug substance or any component of the drug product which is not the chemical entity defined as the drug substance or an excipient in the drug product.”
Impurity Profiling is a group of analyticalactivities for detection, isolation identification/structure elucidation, Quantitative determination of organic and inorganic impurities and residual solvents in bulk drugs &pharmaceutical formulations.
[adsense:468x15:2204050025]
CLASSIFICATION OF IMPURITIES
Impurities can be classified into the following categories:[2]
- Organic Impurities (process and drug related).
- Inorganic impurities.
- Residual solvents.
Organic impurities
It can arise during the manufacturing process and/or storage of the new drug substance. This organic impurities can be identified or unidentified, volatile or nonvolatile and also include -
- Starting materials
- By-products
- Intermediates
- Degradation products
- Reagents, ligands and catalysts
Inorganic Impurities
Inorganic impurities are usually detected and quantified using Pharmacopeial or other appropriate principles. Carryover of catalysts to the drug substance should be evaluated throughout development. Thesekinds of impurities can result from the manufacturing progression. Theseare normally known and identified and include -
- Reagents, ligands and catalysts
- Heavy metals or other residual metals
- Inorganic salts.
- Other materials (e.g., filter aids, charcoal)
Residual Solvents
Solvents are inorganic or organic liquids used as vehicles for the preparation of solutions or suspensions in the synthesis of a new drug substance. The control of residual solvents used in the manufacturing process for the drug substance should be discussed. Acceptance criteria must be based onPharmacopeial standards, or ICH (Q3C) guidelines or known safety data, depends on the dose, duration of treatment, and route of administration.[3] Depending on the possible risk to human health, residual solvents are divided into three classes.
Table No. 1: Classification of residual solvents
|
Solvent |
Risk assessment |
Example |
|
Class I |
Solvents to be avoided |
Benzene (2ppm), Carbon tetrachloride (4ppm), Methylene chloride (600ppm), Methanol (3000ppm), Pyridine (200ppm), Toluene(890ppm) |
|
Class II |
Solvents to be limited |
N, N- dimethyl formamide (880ppm), acetonitrile (410ppm) |
|
Class III |
solvents with low toxic potential |
Acetic acid, Ethanol, Acetone has permitted daily exposure of ≤50mg/day. |
Limits for impurities:
According to the ICH guidelines on impurities in new drug products, identification of impurities below 0.1% level is not measured to be necessary, unless otherwise potential impurities are expected to be unusually potent ortoxic.[3] According to the ICH, the maximum daily dose qualification threshold to be considered is as follows;
Table No. 2: Drug substance impurities thresholds
|
Maximum Daily Dosea |
Reporting Thresholdb,c |
Identification Thresholdc |
Qualification Threshold |
|
≤2 gm/day |
0.05% |
0.1% or 1.0mg /day intake(whichever is lower ) |
0.15% or1.0mg/day intake whichever is lower |
|
>2 gm/day |
0.03% |
0.05% |
0.05% |
a. The amount of drug substance administered per day.
b. Higher reporting thresholds shall be scientifically justified.
c. Lower thresholds can be appropriate if the impurity is unusually toxic.
SOURCES OF IMPURITIES[4,5,6]
From the earlier discussion, it is clear that impurities can originate from several sources; such as;
· Crystallization-related impurities
· Stereochemistry-related impurities
· Impurities arising during storag
· Method related impurity
· Residual solvents
· Synthetic intermediates and by-products
· Functional group-related typical degradation
· Mutual interaction amongst ingredients
Crystallization-related impurities
As per the regulations laid down by the regulatory authorities, a pharmaceutical industry has to take strong enough interest on crystallization related impurities. The nature of structure adopted by a given compound upon crystallization can exert a profound effect on the solid - state properties of that system. Polymorphism of a substance exist in more than one crystalline form whereas, when the substance different crystal packing arrangements with a different elemental composition; the phenomenon is known as Solvatomorphism.
Stereochemistry-related impurities
It is of supreme importance to look for stereochemistry related compounds, that is those compound have similar chemical structure but different spatial orientation; these compound can be considered as impurities. Chiral molecules are frequently called as impurities. The single enantiomeric form of chiral drug may have better pharmacological action and broad therapeutic index.Single isomeric form of drug that are marketed include esomeprazole (S - omeprazole), levabuterol (R- albuterol) etc. The undesired chiral forms of drug are considered as impurity.
Impurities arising during storage
Upon storage or shipment number of impurities can generate in drug products. So it is must and soul to carry out stability studies to predict, evaluate, isolate and ensure the drug product safety. The degradation of penicillin and cephalosporin is well known example of degradation of drug products.
Method related impurity
Sometime a drug produce impurity based upon the formulation of the particular drug product, a well known example is formation of 1-(2, 6-dichlorophenyl) indolin-2-one in the diclofenac sodium ampoules when autoclave under 123±2ºC, that enforce the intramolecular cyclic reaction of diclofenac sodium forming indolinone derivative and sodium hydroxide. Here the formation of this impurity has been found to depend on initial pH of the formulation.
Residual solvents
Residual solvents are organic volatile chemicals used during the manufacturing process or may generate during production. Residual solvents are potentially detrimental substances. They may also affect physicochemical properties of the bulk drug substances such as crystalline of bulk drug, which in turn may affect the dissolution properties, odour and colour changes in finished products. The details of residual solvents as per ICH guideline mentioned above.
Synthetic intermediates and by-products
Impurities in pharmaceuticals or a new chemical entity (NCE) can originate during the synthetic process from raw materials, intermediates and/or by –products. Sometime solvents used for synthesis also contain impurities than traces level. Solvents used in synthesis may produce unwanted impurity and can react with number of chemicals used in the synthesis to produce impurities. By-products from the side reactions are among the most common process impurities in drugs. By-products can be formed through a variety of side reactions, such as incomplete reaction, overreaction, isomerisation, dimerization, and rearrangement, unwanted reactions between starting materials or intermediates with chemical reagents or catalysts.
Functional group-related typical degradation
Functional group-related typical degradation are
- Oxidative degradation
- Ester hydrolysis
- Hydrolysis
- Photolytic cleavage
- Decarboxylation
Oxidative degradationof drugs like Methotrexate, hydrocortisone, hydroxyl group directly bonded to an aromatic ring (viz phenol derivative such as catecholamine’s and morphine), conjugated dienes (viz vitamin A and unsaturated free fatty acids), and heterocyclic aromatic rings, nitroso and nitro derivatives, aldehydes especially susceptible to oxidative degradation.
Ester hydrolysis included the following examples: Aspirin, Benzocaine, ethyl paraben, Cefodoxime proxitil.
Hydrolysis is very natural occurrence for the ester type of drugs, especially in liquid dosage forms. Examples include benzyl penicillin, barbital, chloramphenicol, chlordaizepoxide, oxazpam and lincomycin.
Photolytic cleavage includes following examples:Ergometrine, Nifedipine, sodium nitroprusside, riboflavin and phenothiazines are very labile to photo-oxidation. In susceptible compounds,photochemical energy creates free radical intermediates, which can disseminate chain reactions. Generally compounds will degrade as solutions when exposed to high energy UV exposure. Fluoroquinolones antibiotics are found to be susceptible to photolytic cleavage. Example in ciprofloxacin eye drops preparation (0.3%), sunlightinduces photo cleavage reaction producing ethylenediamine analog of ciprofloxacin.
Decarboxylation includesdissolved carboxylic acids, such as p-amino salicylicacid; lose carbon dioxide from the carboxylgroupwhen heated. Decarboxylation too occurred in the case of photoreaction of rufloxacin.
Mutual interaction amongst ingredients
The majority of vitamins are very labile and upon aging they construct a problem of instability in different dosage forms, especially in liquid dosage forms. Degradation of vitamins does not give any toxic impurities; though potency of active ingredients drops below Pharmacopeial specifications. Because of mutual interaction, the presence of nicotinamide in a formulation containing four vitamins (nicotinamide, pyridoxine, riboflavin, and thiamine) can cause the degradation of thiamine to a sub-standard level within a one year shelf life of vitamin B-complex injections. The marketed samples of vitamin B-complex injections were found to have a pH range of 2.8 - 4.0. A custom-made formulation with simple distilledwater and a typical formulated vehicle including disodium edetate and benzyl alcohol were investigated and similar mutual interactions causing degradation were observed.
Qualification of impurities
Qualification is the process of acquiring and evaluating data that establishes the biological safety of an individual degradation product or a given degradation profile at the level(s) specified. The stage of any degradation product presents in a new drug product that has been tolerably tested in safety and/or clinical studies would be considered qualified[4].An impurity is considered qualified when it meets one or more of the following conditions:
- When the observed level and proposed acceptance standard for the impurity do not exceed the level observed in an FDA approved human drug product.
- When the impurity is a significant metabolite to the drug substance.
- When the observed level and the proposed acceptance standard for the impurity are adequately justified by the scientific literature.
When the observed level and proposed acceptance criterion for the impurity do not exceed the level that has been adequately evaluated in comparative in vitro genotoxicity studies.
Higher or lower thresholds for qualification of degradation products can be appropriate for some individual new drug products based on scientific rationale and level of concern, including drug class effects and clinical experience.
ANALYTICAL METHODS FOR IDENTIFICATION OF IMPURITIES[7,8]:
The impurities can be identified by following different methods like
- Reference standard method
- Spectroscopic method
- Separation method
- Isolation method
- Characterization method
Reference standard method
The main purpose of this method is to afford clarity on the whole life cycle, qualification and control of reference standards used in development and control of new drugs is very important. As because the reference standards provides the basic information for evaluating process and product performance of drug substances, drug products, impurities, degradation products, starting materials, intermediates, and excipients.
Spectroscopic methods
The UV, IR, MS, NMR and Raman spectroscopic methods are abundantly used for the identification of impurities. Now a day’s ICP MS also play a vital role for the identification of impurities. And it has wide choice throughout the different regulatory authority.
Separation methods
The separation method includes chromatographic techniques like TLC, HPTLC, HPLC, Gas Chromatography (GC), Supercritical Fluid Chromatography (SFC), Electrophoresis techniques like Capillary electrophoresis, Gel permeation chromatography etc.
Isolation methods
Number of methods can be used for isolation and characterization of impurities. But the application of any method depends on the nature of impurity (i.e.) itsstructure, physicochemical properties and availability. Predominantly the chromatographic techniques are used for isolation of impurities along with non-chromatographic techniques are also rarely used. The following methods are generally used:
- Solid-phase extraction methods
- Liquid-liquid extraction methods
- Accelerated solvent extraction methods
- Column chromatography
- Flash chromatography
- TLC
- GC
- HPLC
- HPTLC
- Capillary electrophoresis (CE)
- Supercritical fluid chromatography (SFC).
Characterization method
Highly sophisticated instrumentation, such asMS attached to a GC or HPLC, are inevitable tools in the identification of minor components (drugs, impurities, degradation products, metabolites) in various matrices. For characterization of impurities, different techniques are used; which are as follows;
- HPLC-UV Studies
- HPLC-MS Studies
- GC-MS Studies
- TLC-MS Studies
- CE-MS Studies
- MEKC-MS and CEC-MS Studies
- HPLC-NMR Studies
Fig No. 1: Systemic approach for impurity determination
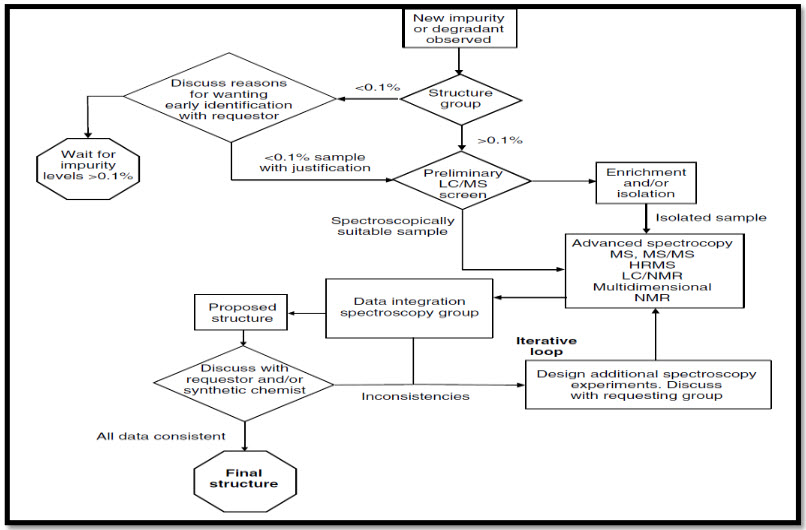
Table No. 3: Some examples of drugs and their Impurities
|
Drug |
Impurity |
Analytical Method |
|
Atropine sulphate |
Apo atropine |
Ultra violet spectroscopy |
|
Cloxacillin |
N,N dimethyl aniline |
Gas chromatography |
|
Dextrose
|
5 hydroxy methyl fulfural |
Ultra violet spectroscopy |
|
Diclofenac Sodium |
1-(2,6-dichlorophenyl) indolin -2-one |
Liquid chromatography |
|
Doxorubicin hydrochloride |
Acetone and ethanol |
Gas chromatography |
|
Ethambutol hydrochloride |
2 – amino butanol |
Thin Layer Chromatography |
|
Framycetin sulphate |
Neamycin |
Ultra violet spectroscopy |
|
Methamphetamine |
1,2-dimethyl-3-phenylaziridine, ephedrine,methyl ephedrine, N formyl methamphetamine, Nacetyl methamphetamine, N formylphedrine, Nacetyl ephedrine, N,Odiacetylephedrine, methametamine dimmer |
Gas chromatography |
|
Mercaptopurine |
Hypoxanthine |
Ultra violet spectroscopy |
Validation of analytical methods
The validation process involves confirmation or establishing a developed method by laboratory studies, procedures, systems, which can give accurate and reproducible result for an intended analytical technique[8]. Results from method validation can be used to review the quality, reliability and consistency ofresults and it is an integral part of good analytical practice.
Table No. 4:Validation parameter for impurities present in drug substances
|
Types of analytical procedure characteristic |
Identification |
Testing for impurities |
||
|
Quantitative |
Limit |
|||
|
Linearity |
- |
+ |
- |
|
|
Range |
- |
+ |
- |
|
|
Accuracy |
- |
+ |
- |
|
|
Precision
|
Repeatability |
- |
+ |
- |
|
Intermediate |
- |
+(a) |
- |
|
|
Specificity(b) |
+ |
+ |
+ |
|
|
Limit of Detection(LOD) |
- |
-(c) |
+ |
|
|
Limit of Quantitation (LOD) |
- |
+ |
- |
|
- Signifies that this characteristic is not normally evaluated.
+ Signifies that this characteristic is normally evaluated.
(a) In case where reproducibility has been performed, intermediate precision is not needed.
(b) Lack of specificity of one analytical procedure could be compensated by other supporting analytical procedure.
(c) May be needed in some cases.
CONCLUSION
This review provides a perception on impurities in drug substance and drug products. Various regulatory authorities such as USFDA, MHRA, TGAand ICH already given emphasize in impurity profiling and governed such limit and regulation on this. So it is very important to health care authorities give more attention on impurities present on drug product as because a little amount of impurities present in drug products can alter the biological as well as therapeutic efficacy. From the safety point of view also it is essential to do evaluation, isolation and detection of impurities.This article provides the important information about the impurities types, source and its classification, various techniques of isolation and characterization, analytical techniques for the determination, qualification of impurities and critical factors to be considered while preparation of the bulk drugs. Isolation and characterization of impurities is obligatory for acquiring and evaluating data that establishes biological safety which reveals the need and scope of impurity profiling of drugs in pharmaceuticals.
REFERENCES
1. Guidance for Industry, ANDAs: Impurities in DrugProducts, U.S. Department of Health and HumanServices, Food and Drug Administration, Centre forDrug Evaluation and Research, August 2005(http://www.fda.gov/cder/guidance/6423dftrev1.htm).
2. Guidance for Industry, ANDAs: Impurities in Drug Substances, U.S. Department of Health and HumanServices, Food and Drug Administration, Centre for Drug Evaluation and Research, January 2005(http://www.fda.gov/cder/guidance/6422dft.htm).
3. International Conferences on Harmonization, Draft revised Guidance on Impurities in new drug substances Q3A(R2)., 25 October 2006
4. U.S. Food and Drug Administration. Guidance for Industry, Q3A Impurities in New Drug Substances. February 2003.
5. Ahuja Satinder. Impurities Evaluation of Pharmaceuticals. Ed., New York; Marcel Dekker. 1998; 15-18.
6. Parimoo P. A Text Book of Pharmaceutical Analysis, CBS publishers and distributors, NewDelhi. 1998; 20-21.
7. S. J. Ingale et al. Advance approaches for the impurity profiling of pharmaceutical drugs: A review. Int. J. of Pharm. & Life Sci 2011;2(7);955-962.
8. Pradeep Patil, Dr. Vaidya. Overview on Impurity Profiling. Int. J. For Pharm Res Sch.,2013;2(2):54-65.
9. Roy J. Pharmaceutical impurities-A mini review. AAPS Pharm Sic Tech 2002;3(2);1-11.
10. The Nature and Origin of the Impurities in Drug Substances (1.2), Sdndor Ggrgg Chapter 1. 1998;12-13.
11. Residual Solvents, Q3C. Federal Register, 62(247), 2000; 67377.
REFERENCE ID: PHARMATUTOR-ART-2373
|
PharmaTutor (Print-ISSN: 2394 - 6679; e-ISSN: 2347 - 7881) Volume 3, Issue 11 Received On: 22/07/2015; Accepted On: 02/08/2015; Published On: 01/11/2015How to cite this article: A Chanda, N Ramalakshmi, CN Nalini, S Mahabubi; Impurity profiling an emerging trend in Pharmaceuticals: A Review; PharmaTutor; 2015; 3(11); 29-35 |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






