

About Authors:
Krunal Parikh*, Mr. Maheshkumar Kataria, Jatin Patel, Tarun Patel, Dhiren Shah
Seth G. L. Bihani S.D. College of Technical Education, Institute of Pharmaceutical Sciences and Drug Research,
Sriganganagar, India
*Krunal_2922@yahoo.in
ABSTRACT
Enzyme-linked immunosorbent assays (ELISAs) are plate-based assays designed for detecting and quantifying substances such as peptides, proteins, antibodies and hormones. Other names, such as enzyme immunoassay (EIA), are also used to describe the same technology. In an ELISA, an antigen must be immobilized to a solid surface and then complexed with an antibody that is linked to an enzyme. Detection is accomplished by assessing the conjugated enzyme activity via incubation with a substrate to produce a measureable product. The most crucial element of the detection strategy is a highly specific antibody-antigen interaction. There are three methods of ELISA. Each type of ELISA can be used qualitatively to detect the presence of antibody or antigen. Alternatively, a standard curve based on known concentration of antibody or antigen is prepared, from which the unknown concentration of a sample can be determined.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1462
INTRODUCTION
Enzyme immunoassay, a bioanalytical method incorporatingan antigen–antibody reaction to capture theanalyte of interest and an enzyme reporter system todetect the captured analyte, is one of the most widelyused immunoassay formats. The method is sometimesapplied only qualitatively to indicate the presence ofan antigen in a matrix. However, in the more common.
Quantitative implementation, a calibration (standard) curve is incorporated, from which the concentrationof the analyte in unknown samples is interpolated. Immunoassayshave been widely applied in support ofmedical practice and drug development. However, inrecent years, there has been a decline in the applicationof immunoassays to the quantitation of low-molecularweightxenobiotics, primarily due to the advent ofliquid chromatography–mass spectrometry (LC–MS)methods, which have high sensitivity and specificity. Immunoassaysremain the method of choice for the quantitation ofprotein macromolecules and antibodies in complexmatrices. Another major application of immunoassaysis in the detection and quantitation of biomarkers, whichare evolving to be of pivotal importance in the evaluationof pharmacological, toxicological, and clinical activities ofcandidate drugs1.Immunoassays generally vary in the type of criticalantibody binding reagent or the detection and reportersystems used to monitor the end-point of the bindingreaction.Enzyme immunoassay formatsfall broadly into two categories, namelyheterogeneous and homogeneous. In a heterogeneousassay, at least one key reagent is immobilized on a solidsurface and there is at least one ‘‘washing’’ step beforethe final detection step. In contrast, in a homogeneousassay, all reagents are in solution together and there isno ‘‘washing’’ step prior to signal generation anddetection. Both categories of assay include formatsdescribed as competitive and non-competitive. In acompetitive assay, there is direct competition betweenthe labeled and the unlabeled antigen (analyte orligand) in solution or, in some cases, between immobilizedand soluble antigen for a limited number of antibodybinding sites. In non-competitive assays, antibodybinding sites to capture and detect the antigen are notlimiting because the antigen is incubated with excesscapture antibody and enzyme-labeled detection antibody.An example of a competitive homogeneousassay format is the enzyme-multiplied immunoassay(EMIT) system, in which enzyme-labeled antigencompetes directly in solution with unlabeled antigenin the biological sample (or calibration standard andquality control samples) for a limited number of antibodybinding sites. The reaction endpoint is detectedand quantitated spectrophotometrically without anyintervening wash steps. Enzyme-linked immunosorbent assay(ELISA) is an example of a heterogenous noncompetitiveimmunoassay2.
[adsense:468x15:2204050025]
ELISA- (ENZYME LINKED IMMUNOSORBENT ASSAY)
Enzyme immunoassay (EIA) and enzyme-linked immunosorbentassay (ELISA) have become household namesfor medical laboratories, manufacturers of in vitro diagnostic products, regulatory bodies, external qualityassessment and proficiency-testing organizations3. It is a biochemical technique used mainly in immunology to detect the presence of an antibody or an antigen in a sample4. In principle, the method is similar to that of the RIA which involves competitive binding, it does not involve in use of any radio-labelled material and the sensitivity of the method is equally good 0.0001-0.001 µg/ml. the method is safe and cheaper as compared to RIA. Here the enzyme conjugated to an antibody reacts with a colorless substrate to generate a colored reaction product. The color is measured spectrophotometrically. The most commonly used enzymes are horseradish peroxidase, alkaline phosphatase, p-nitrophenyl phosphatase β galactosidase5. These enzymes have been used to link to the antibody or antigen molecule as may be the case. When mixed with a suitable substrate each of these enzymes generates a colored reaction product6. ELISA procedure are popular primarily because they require little interpretive skill to read the results tend to be clearly positive or clearly negative7.
HISTORY
Before the development of the EIA/ELISA, the only option for conducting an immunoassay was radioimmunoassay, a technique using radioactively -labeled antigens or antibodies. In radioimmunoassay, the radioactivity provides the signal which indicates whether a specific antigen or antibody is present in the sample. Radioimmunoassay was first described in a paper by Rosalyn Sussman Yalowand Solomon Bersonpublished in 1960.
Because radioactivity poses a potential health threat, a safer alternative was sought. A suitable alternative to radioimmunoassay would substitute a non-radioactive signal in place of the radioactive signal. When enzymes (such as peroxidase) react with appropriate substrates (such as ABTSor 3, 3’, 5, 5’-Tetramethylbenzidine), this causes a change in color, which is used as a signal. However, the signal has to be associated with the presence of antibody or antigen, which is why the enzyme has to be linked to an appropriate antibody. This linking process was independently developed by Stratis Avrameas and G.B. Pierce. Since it is necessary to remove any unbound antibody or antigen by washing, the antibody or antigen has to be fixed to the surface of the container, i.e. the immunosorbent has to be prepared. A technique to accomplish this was published by Wide and Jerker Porathin 19664.
In 1971, Peter Perlmann and Eva Engvallat Stockholm University in Sweden, and Anton Schuurs and Bauke van Weemen in The Netherlands, independently published papers which synthesized this knowledge into methods to perform EIA/ELISA3.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
NUMEROUS VARIANTS OF ELISA
A number of variation ELISA have been developed, allowing qualitative detection or quantitative measurement of either antigen or antibody. Each type of ELISA can be used qualitatively to detect the presence of antibody or antigen. Alternatively, a standard curve based on known concentration of antibody or antigen is prepared, from which the unknown concentration of a sample can be determined.
1. Indirect ELISA
Antibody can be detected or quantitatively determined with an indirect ELISA. Serum or some other sample containing primary antibody (Ab1)is added to an antigen- coated microtiter well and allowed to react with the antigen attached to the well. After any free Ab1 is washed away, the presence of antibody bound to the antigen is determined by adding an enzyme- conjugated secondary anti- isotope antibody (Ab2), which binds to the primary antibody. Any free Ab2 then is washed away, and a substrate for the enzyme is added. The amount of colored reaction product that forms is measured by specialized spectrophotometric plate readers, which can measure the absorbance of all of the wells of a 96- well plate in seconds8.
Indirect ELISA is the method of choice to detect the presence of serum antibodies against human immunodeficiency virus (HIV), the causative agent of AIDS. For detection of anti- HIV-1 and anti- HIV-2 antibodies in the patientserum. The well of the polystyrene microtitre plate are coated with purified HIV-1 and HIV-2 antigens or synthetic peptides representing immunodominant epitopes of HIV-1 and HIV-2, which constitutes the solid phase antigens. Diluted test serum or plasma sample is added to such a well and incubated. If antibodies specific for HIV-1 and/or HIV-2 is/are present in the test sample they will form stable complexes with antigens coated on the well. Well is then washed and a conjugate of goat anti-human immunoglobin, which has been labelled with the enzyme horse-radish peroxidase, is added. If the antigen- antibody complex is present, the peroxidase conjugate will bind to the complex and remains in the well is removed by washing and the presence of enzyme immobilized on the complexes is shown by incubation in the presence of enzyme substrate (ortho-phenylene-diamine dihydrochloride solution). Incubation with enzyme substrate produces a yellow-orange colour in the test well. If the sample contains no anti-HIV-1 and/or anti-HIV-2, then the labeled antibody cannot be found and no colour develops. The absorbance value of each well is read by an ELISA plate read at wavelength of 492±2 nm9. This method diagrammatically shown in fig. 1
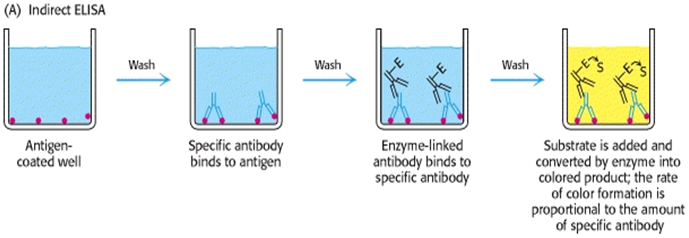
1) indirect ELISA method
2. Sandwich ELISA
One of the most useful of the immunoassays is the two antibody “sandwich” ELISA. This assayis used to determine the antigen concentration in unknown samples. This ELISA is fast andaccurate, and if a purified antigen standard is available, the assay can determine the absoluteamount of antigen in an unknown sample. The sandwich ELISA requires two antibodies thatbind to epitopes that do not overlap on the antigen. This can be accomplished with either twomonoclonal antibodies that recognize discrete sites or one batch of affinity-purified polyclonalantibodies. Antigen can be detected or measured by a sandwich ELISA.
In this technique, the antibody (rather than the antigen) is immobilized on a microtiter well. A sample containing antigen is added and allowed to react with the immobilized antibody. After the well is washed, a second enzyme- linked antibody specific for a different epitopes on the antigen is added and allowed to react with the bound antigen. After any free second antibody is removed by washing, substrate is added, and the colored reaction product is measured8. This method diagrammatically shown in fig. 2
The Sensitivity of the Sandwich ELISA is Dependent on Four Factors
1. The number of molecules of the first antibody that are bound to the solid phase.
2. The avidity of the first antibody for the antigen.
3. The avidity of the second antibody for the antigen.
4. The specific activity of the second antibody.
The amount of the capture antibody that is bound to the solid phase can be adjusted easily bydilution or concentration of the antibody solution. The avidity of the antibodies for the antigencan only be altered by substitution with other antibodies. The specific activity of the secondantibody is determined by the number and type of labeled moieties it contains10.
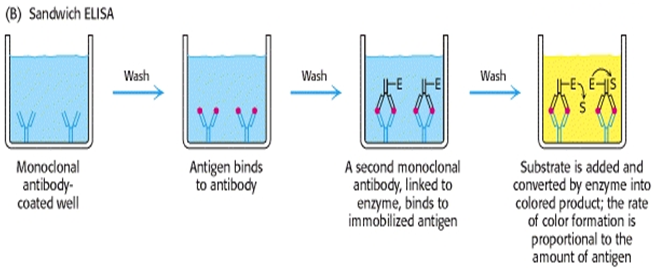
2) sandwich ELISA method
3. Competitive ELISA
When two “matched pair” antibodies are not available for your target, another option is the competitive ELISA. Another advantage to the competitive ELISA is that non-purified primary antibodies may be used. Another variation for measuring amounts of antigen is competitive ELISA10.
In this technique, antibody is first incubated in solution with a sample containing antigen. The antigen- antibody mixture is then added to an antigen-coated microtiter well. The more antigens present in the sample, the less free antibody will be available to bind to the antigen-coated well. Addition of an enzyme conjugated secondary antibody (Ab2) specific for the isotope of the primary antibody can be used to determine the amount of primary antibody bound to the well in an indirect ELISA8. This method diagrammatically shown in fig. 3
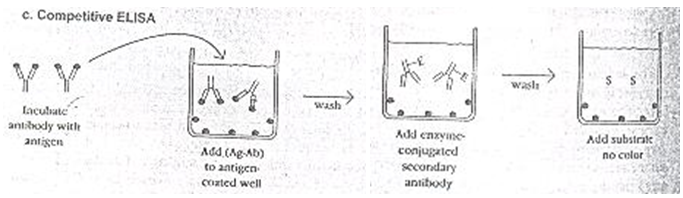
3) competitive ELISA method
4. Reverse ELISA
A new technique uses a solid phase made up of an immunosorbent polystyrene rod with 4-12 protruding gives. The entire device is immersed in a test tube containing the collected sample and the following steps (washing, incubation in conjugate and incubation in chromogenous) are carried out by dipping the gives in microwells of standard micro plates pre-filled with reagents.
The advantages of this technique are as follows:
1. The gives can each be sensitized to a different reagent, allowing the simultaneous detection of different antibodies and different antigens for multi-target assays;
2. The sample volume can be increased to improve the test sensitivity in clinical (saliva, urine), food (bulk milk, pooled eggs) and environmental (water) samples;
3. One give is left unsensitized to measure the non-specific reactions of the sample;
4. The use of laboratory supplies for dispensing sample aliquots, washing solution and reagents in microwells is not required, facilitating ready-to-use lab-kits and on-site kits4.
The advantages of this technique are as follows:
- The gives can each be sensitized to a different reagent, allowing the simultaneous detection of different antibodies and different antigens for multi-target assays;
- The sample volume can be increased to improve the test sensitivity in clinical (saliva, urine), food (bulk milk, pooled eggs) and environmental (water) samples;
- One give is left unsensitized to measure the non-specific reactions of the sample;
-
The use of laboratory supplies for dispensing sample aliquots, washing solution and reagents in microwells is not required, facilitating ready-to-use lab-kits and on-site kits4.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
FACTORS THAT INFLUENCE ASSAY PERFORMANCE
- The greatest impact of the behavior of the assay is typically based on the antibody pair itself. If the pair is a “good” match for the desired analyte, other unwanted factors are greatly minimized. If the pair is a “poor” match with lower affinity for the desired analyte, it may take a lot of time and creativity to achieve acceptable parameters.
- Non-specific binding can cause high background values and poor recovery and linearity, which can often be controlled with IgG antibodies or serum.
- False positive values can occur on Non-specific binding of samples to one or both of the antibodies present which is often detected when performing spiked recovery and linearity testing. In the ELISA format, these samples can typically be controlled by the use of IgG antibodies and/or heterophilic blockers.
- Hook Effect may give incorrect sample values for highly concentrated samples and can usually be detected during linearity studies.
VALIDATION OF ELISA ASSAYS
1. Spiked recovery tests are performed using the blank, low standard, medium standard and high standard points spiked into at least 8 different samples in duplicate. This test should be repeated at least two times to determine if results are reproducible and within acceptable criteria for the coefficient of variation (CV). Acceptable criteria are CVs of 80–120%.
2. Linearity tests are performed on at least 8 different samples in duplicate. At least three dilution of the neat samples should be performed and more if possible. This test should be repeated at least two times to assure that the results are reproducible and acceptable (80–120% CVs).
3. Intra-assay variation is tested by running 8 different samples (of varying concentration) in replicates of ten across the microtiter plate and determining the % CVs of the samples. Acceptable criteria are typically CVs of 80–120%.
4. Inter-assay variation is determined by evaluating at least 8 samples (of varying concentration) in duplicate on at least three different microtiter plates on different days using the same reagent lots. The % CVs are then calculated. Acceptable criteria is typically CVs of 80–120%.
5. The microtiter plates coated with the capture antibody must also be validated by running the blank, low, and high standard points in replicates of 32, on a minimum of three plates, and the % CVs calculated. Acceptable criteria is typically CVs of =10%.
6. The sensitivity of the assay is determined by evaluating the blank and the lowest standard point in replicates of at least 20. The following calculation is used to determine the sensitivity of the assay:
7. Further specificity of the assay is evaluated by running standards of several similar analytes of available species in the assay to determine if any similar analytes can be detected by the assay.
Sensitivity = Background Mean x low std. Mean
---------------------------
low Std.- Mean Background Mean
8. If available, the sample values are compared to sample values in the most popular competitor kit or our own Millipore RIA kit to determine the correlation of sample values.
9. Compare serum vs. plasma values and determine the correlation. Also evaluate if assay can be performed on cell extracts or cell culture media11.
APPLICATIONS
Because the ELISA can be performed to evaluate either the presence of antigen or the presence of antibody in a sample, it is a useful tool for determining serum antibody concentrations (such as with the HIV testor West Nile Virus). It has also found applications in the food industry in detecting potential food allergens such as milk, peanuts, walnuts, almonds, and eggs.ELISA can also be used in toxicology as a rapid presumptive screen for certain classes of drugs. ELISA provides a means to quantitatively measure extremely small amount of proteins in biological fluids and serves as a tool for analyzing specific protein during purification12.
The ELISA was the first screening test widely used for HIV because of its high sensitivity. In an ELISA, a person's serum is diluted 400-fold and applied to a plate to which HIV antigens are attached. If antibodies to HIV are present in the serum, they may bind to these HIV antigens. The plate is then washed to remove all other components of the serum. A specially prepared "secondary antibody" — an antibody that binds to other antibodies — is then applied to the plate, followed by another wash. This secondary antibody is chemically linked in advance to an enzyme. Thus, the plate will contain enzyme in proportion to the amount of secondary antibody bound to the plate. A substrate for the enzyme is applied, and catalysis by the enzyme leads to a change in color or fluorescence. ELISA results are reported as a number; the most controversial aspect of this test is determining the "cut-off" point between a positive and negative result.
A cut-off point may be determined by comparing it with a known standard. If an ELISA test is used for drug screening at workplace, a cut-off concentration, 50 ng/ml, for example, is established, and a sample will be prepared which contains the standard concentration of analyte. Unknowns that generate a signal that is stronger than the known sample are "positive". Those that generate weaker signal are "negative."
ELISA can also be used to determine the level of antibodies in faecal content...specifically the direct method.
ELISA test are utilized to detect substances that have antigenic properties, primarily proteins (as opposed to small molecules and ions such as glucose and potassium). Some of these include hormones, bacterial antigens and antibodies. ELISA methods are fundamental tools in the pharmaceutical industry with applications in drug discovery, animal studies, and clinical trials13.
There are variations of this test, but the most basic consists of an antibody attached to a solid surface. This antibody has affinity for (will latch on to) the substance of interest, for example, human chorionic gonadotropin (HGC), the commonly measured protein which indicates pregnancy. A mixture of purified HCG linked (coupled) to an enzyme and the test sample (blood, urine, etc) are added to the test system. If no HCG is present in the test sample, then only HCG with linked enzyme will bind. The more HCG which is present in the test sample, the less enzyme linked HCG will bind. The substance the enzyme acts on is then added, and the amount of product measured in some way, such as a change in color of the solution14.
ELISA tests are generally highly sensitive and highly specific and less expensive technique used in serology to detect antigens or antibodies15. They have the added advantages of not needing radioisotopes or a radiation-counting apparatusELISA assays are widely applied in clinical laboratory testing16.
|
Direct Detection |
Advantages |
· Quick because only one antibody and fewer steps are used. · Cross-reactivity of secondary antibody is eliminated. |
|
Disadvantages |
· Immunoreactivity of the primary antibody might be adversely affected by labeling with enzymes or tags. · Labeling primary antibodies for each specific ELISA system is time-consuming and expensive. · No flexibility in choice of primary antibody label from one experiment to another. · Minimal signal amplification. |
|
|
Indirect Detection |
Advantages |
· A wide variety of labeled secondary antibodies are available commercially. · Versatile because many primary antibodies can be made in one species and the same labeled secondary antibody can be used for detection. · Maximum immunoreactivity of the primary antibody is retained because it is not labeled. · Sensitivity is increased because each primary antibody contains several epitopes that can be bound by the labeled secondary antibody, allowing for signal amplification. · Different visualization markers can be used with the same primary antibody. |
|
Disadvantages |
· Cross-reactivity might occur with the secondary antibody, resulting in nonspecific signal. · An extra incubation step is required in the procedure. |
COMPARISON OF DIRECT AND INDIRECT ELISA DETECTION METHODS17
BIBLIOGRAPHY
1. Prescott L.M.etal “ Microbiology” Third edition International edition 2003 printed in Singapore PP 778-779
2. James Swarbrick, James C. Boylan “Encyclopedia of Pharmaceutical technology” second edition, volume 2, Marcel Dekker Series 2002 PP- 70-79,994-1013.
3. Lequin R.M. “Enzyme Immunoassay (EIA)/ Enzyme- linked Immunosorbent Assay (ELISA) American Association for Clinical Chemistry, Inc.© 2005 First published September 22, 2005; Available from
clinchem.org/cgi/content/full/51/12/2415
4. ELISA, Available from
en.wikipedia.org/wiki/ELISA
5. Parija S.C. “Textbook of practical microbiology” first ediion 2007 Ahuja publication house delhi PP 138, 141
6. Dr. Vyas S.P.; Dr.Dixit V.K.” Pharmaceutical biotechnology” Reprint 2004 CBS publishers & Distributors New Delhi PP 138, 477
7. Tortora G.J.; Funke B.R. “Microbiology” An introduction fifth edition 2005 publlished bypearson education (Singapore) Pvt. Ltd. Delhi PP 321, 387, 552, 553, 554
8. Goldsby R.A. etal; “Immunology” fifth edition 2003 published by W.H. Freeman & company Newyork PP 148-151
9. Arora D.R. “Text book of microbiology” first edition1999 CBS publishers & Distributors New Delhi PP 112,113,242,273
10. Introduction to Antibodies - Enzyme-LinkedImmunosorbent Assay (ELISA), Available from
biosupply.co.uk/doc.php?id=2601
11. Enzyme Linked Immunosorbent Assay (ELISA), Millipore, Available from
millipore.com/immunodetection/id3/elisa
12. CrommelinDaan J.A.; Sindelar R.D. “ Pharmaceutical biotechnology” an introduction for pharmacist and pharmaceutical scientist second edition published by taylor&francis London PP 42-44
13. S.L.Fosscecoetal; “Exploring Enzyme-Linked Immunosorbent Assay (ELISA) Data with theSAS® Analyst Application”, (SAS Institute Inc., Cary, NC), 1-8 Available from
support.sas.com/rnd/app/papers/analystelisa.pdf
14. Medicine net.com, Definition of Enzyme- linked immunosorbent assay (ELISA) (Review on 13 April 1999) Available from
medterms.com/script/main/art.asp?articlekey=9100
15. Reddy K.R “ Microbiology and parasitology” first edition 2002 published by Divyesh A. Kothari Paras Medical publisher Hyderabad; PP 240, 462
16. Tierney L.M.; Mcphee S.J. “Current medical Diagnosis and treatment“ International edition 1994 Published by Appleton and lange USA PP 650.
17. Thermo Scientific, protein method library (Overview of Elisa) Thermo Fisher Scientific Inc.© 2011 Available from
piercenet.com/browse.cfm?fldID=F88ADEC9-1B43-4585-922E-836FE09D8403
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE







