

.JPG) About Author:
About Author:
Swapna.G*
Department of pharmaceutical Pharmaceutical & Quality Assurance,
Nirmala College of Pharmacy, Mangalagiri, Atmakuru, Guntur -522 203.
*swapna.goday.gs@gmail.com
Abstract
A simple, precise and accurate reversed phase liquid chromatographic method has been developed for the assay of azithromycin in powder for oral suspension. The chromatographic separation was achieved on a Asahipak ODP 40 E(250 mm × 4.6 mm, 5 μm) analytical column. A mixture of methanol–ammonium dihydrogen phosphate (0.05M) (30:70, v/v) (pH 9.0) was used as the mobile phase, at a flow rate of 1.5 mLmin-1 and detector wavelength at 210 nm. The retention time of azithromycin was found to be at 8.0 min. The validation of the proposed method was carried out for specificity, linearity, accuracy, precision, robustness and stability indicating assay. The linear dynamic range is from 382–1208 μgmL-1 for azithromycin. The percentage recovery obtained for azithromycin is 101.0%. The developed method can be used for pharmaceutical dosage form and in process testing.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-1711
Introduction
Azithromycin [9-de-oxy-9a-aza-9a-methyl-9a-homoerythromycin A dihydrate] is an azalide, a subclass of macrolide antibiotics as shown in Fig.1. It is derived from erythromycin; however it differs chemically from erythromycin in that a methyl substituted nitrogen atom is incorporated into the lactone ring, thus making the lactone ring 15 membered. Azithromycin powder for oral suspension is indicated for the treatment of the following infections when caused by microorganisms sensitive to azithromycin particularly those of the respiratory tract, such as pharyngitis / tonsilities, uncomplicated skin and soft tissue infections. It is also effective against certain urinary tract infections and veneral diseases, such as non-gonococcal urithrities, Chlamydia,gonorrhea and cervicities [1-2]. The assay is performed for azithromycin in powder for oral suspension 200mg/5mL. Assays reported in the literature for the determination of azithromycin in biological fluids include HPLC using atmospheric pressure chemical ionization [3], columetric and amperometric detection [4], and fluorescence detection [5],and microbiological diffusion method on an 8 x 8 Latin Square to find out which of the test microorganisms used show the highest sensitivity [6].The methods reported in literature for the determination of azithromycin is HPLC, using electrochemical detection [7,8] or UV detector [9]. The present manuscript describes a simple, rapid, precise and accurate isocratic reversed phase HPLC method for assay, impurity interference and stability indicating assay of azithromycin in powder for oral suspension dosage form.The developed method was validated as per the International Conference on Harmonization (ICH) guideline [10].
Experimental
Materials
Azithromycin standard and impurity-F(Azithromycin related compound), impurity-I(N-Demethylazithromycin),impurity-J(Desosaminylazithromycin),impurity-L(AzithromycinN-oxide) was supplied by Aziant Drug Research Solutions,Hyderabad, India. Ammonium dihydrogen phosphate, Hydrogen peroxide, Sodium hydroxide , HPLC methanol, Hydrochloric acid were purchased from E. Merck (India) Ltd. Worli,Mumbai, India. The 0.45µm nylon filters and PVDF filters were purchased from Advanced Micro Devices Pvt.Ltd.Chandigarh, India. MilliQ water was used throughout the experiment.
Equipments
Analysis was performed on a chromatographic system of Agilent 1200 series with variable wave length detector and photodiode array detector. A chromatographic separation was achieved on Asahipak ODP 4E (250 mm × 4.6 mm, 5 μm) analytical column. Data acquisition was made with EZchrom elite software .The peak purity was checked with the photodiode array detector.
Standard preparation
Standard stock solution of azithromycin (0.8 mg mL-1) was prepared in diluent which was a mixture of methanol and water (50:50,v/v). Filtered about 10mL of above solution through 0.45µ nylon syringe filter by discarding first 4 mL of solution.
Standard solution and calibration graph
Standard stock solution of azithromycin (106.45 mg/mL) was prepared in diluent which was a mixture of methanol and water (50:50,v/v). To study the linearity range serial dilutions were made by adding this standard stock solution in the range of 50 to150% (382-1208 mg mL-1). A graph was plotted as concentration of drug versus peak area response. It was found to be linear. The system suitability test was performed from five replicate injections of standard solution.
Sample preparation
The azithromycin powder for oral suspension container was tapped initially to loosen the powder. The powder was reconstituted by adding the required quantity of water .The reconstituted container was shaked for approximately 15 minutes. The container was allowed to stand for about 30 minutes. The shaking procedure of the container was repeated for an additional 15 minutes. 6.4 g of the reconstituted suspension was taken in 250 mL volumetric flask through 10mL syringe. Made upto the mark with diluent. Filtered about 10mL of above solution through 0.45µm nylon syringe filter by discarding first 4 mL solution.
Method validation
The HPLC method was validated in terms of precision, accuracy and linearity according to ICH guidelines [10]. Assay method precision was determined using six-independent test solutions. The intermediate precision of the assay method was also evaluated using different analyst on different days. The accuracy of the assay method was evaluated by spiking the azithromycin drug substance on placebo in the range of about 50 to 150% level. The sample was extracted as described in Section 2.4 and were analyzed using the developed HPLC method. Linearity test solutions were prepared as described in Section 2.3. The degradation of azithromycin and placebo was performed under different stress conditions (hydrolysis, oxidation,UV,humidity,alkaline, acidic, and thermal stress).To determine the robustness of the method, the final experimental conditions were altered and the results were examined. The flow rate was varied by (±) 0.1 mLmin-.1 The percentage of organic modifier was varied by (±) 5%. Column temperature was varied by (±) 5 °C and pH of mobile phase was varied by (±) 0.1.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Results and discussion
Optimization of the chromatographic conditions
During the analysis of basic drugs like azithromycin one of the well known problem is peak tailing. Since these compounds strongly interact with polar ends of HPLC column packing materials, causing severe peak asymmetry and low separation efficiencies. High purity silica backbone and advances in bonding technology have alleviated the tailing problem of polar compounds in HPLC to a significant extent. During the optimization of the method, different columns (Inertsil C8, 250 mm × 4.6 mm, 5 μm; Zorbax C18 250 mm × 4.6 mm, 5 μm; Symmetry C18 250 mm × 4.6 mm, 5 μm) and two organic solvents (acetonitrile and methanol) were tested. The chromatographic conditions were also optimized by using different buffers like phosphate, acetate and citrate for mobile phase preparation. After a series of screening experiments, it was concluded that phosphate buffers gave better peak shapes than their acetate and citrate counterparts. The chromatographic separation was achieved on a Asahipak ODP 4E (250 mm × 4.6 mm, 5 μm) column, by using a mixture of ammonium dihydrogen phosphate–methanol (0.05M) (30:70, v/v) as mobile phase. Asahipak columns are having high pH (9–13) and temperature (20–60 °C) stability. About mobile phase, due to the lack of other chromophore than the ester group in azithromycin and, therefore, the need to work at a low wavelength (210 nm), methanol was considered as organic solvent instead of acetonitrile . At 30°C column temperature and pH 9.0 of mobile phase, the peak shape of azithromycin was found symmetrical. The flow rate kept was 1.5mL/min to achieve adequate retention time of azithromycin peak (Fig. 1-2)
Validation of method
Specificity
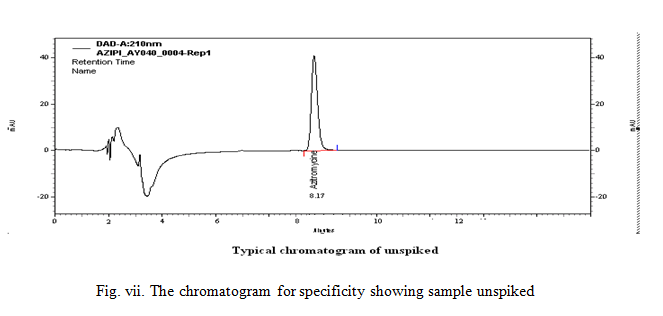
The specificity of the HPLC method is illustrated in (Figs. 3-7) where complete separation of azithromycin was noticed in presence of impurities. In addition there was no interference at analysis with photo diode detector, purity angle was less than purity threshold for the analyte. he retention time of azithromycin in the chromatogram of placebo solution. In peak purity This shows that the peak of analytes was pure and excipients in the formulation did not interfere the analyte
Selectivity
Neither formulation ingredients nor degradation products interfered with quantitation of azithromycin. All samples and placebo were analyzed using the assay chromatographiccondition described. No evidence of interactive degradation products was seen during evaluation. However azithromycin was observed to be susceptible to acidic and oxidative condition. So avoid acidic condition during analysis.Mild degradation was observed in base , water, thermal, and UV degradation.Selectivity was demonstrated showing that azithromycin peak was free of interference of degradation products indicating that the proposed method is stability indicating.
Accuracy
Accuracy of the method was calculated by recovery studies at six levels for 50% and 150% level and three levels for 75%, 100%, and 125%. (Table 1).The mean percentage recovery obtained for azithromycin was found to be in between 99.90 and 101.0% respectively.
Table 1
Results of the recovery analysis of azithromycin
|
S.No. |
% spike level |
Amount added(mg) |
Amount recovered (mg) |
%Recovery |
%Mean Recovery |
% RSD |
|
1. |
50 |
93.71 |
93.60 |
99.9 |
100.2 |
0.4 |
|
2. |
93.91 |
93.63 |
99.7 |
|||
|
3. |
93.79 |
94.32 |
100.6 |
|||
|
4. |
93.72 |
94.18 |
100.5 |
|||
|
5. |
93.65 |
94.04 |
100.4 |
|||
|
6. |
93.62 |
94.00 |
100.4 |
|||
|
1. |
75 |
148.01 |
147.96 |
100.0 |
99.9 |
0.2 |
|
2. |
148.19 |
148.20 |
100.0 |
|||
|
3. |
148.16 |
147.74 |
99.7 |
|||
|
1. |
100 |
198.68 |
199.09 |
100.2 |
100.2 |
0.1 |
|
2. |
198.73 |
199.42 |
100.3 |
|||
|
3. |
198.23 |
198.37 |
100.1 |
|||
|
1. |
125 |
243.69 |
245.84 |
100.9 |
101.0 |
0.2 |
|
2. |
243.75 |
246.56 |
101.2 |
|||
|
3. |
243.57 |
245.87 |
100.9 |
|||
|
1. |
150 |
299.84 |
302.39 |
100.9 |
100.9 |
0.3 |
|
2. |
299.48 |
301.45 |
100.7 |
|||
|
3. |
299.51 |
302.65 |
101.1 |
|||
|
4. |
299.80 |
302.24 |
100.8 |
|||
|
5. |
299.85 |
301.51 |
100.6 |
|||
|
6. |
299.84 |
303.82 |
101.3 |
Precision
The precision of an analytical procedure expresses the closeness of agreement between a series of measurements obtained from multiple sampling of the same homogeneous sample under the prescribed conditions. The system precision is a measure of the method variability that can be expected for a given analyst performing the analysis and was determined by performing five replicate analyses of the same working solution. The relative standard deviation (R.S.D.) obtained for azithromycin was 0.2. (Table2).
Table 2
System suitability parameters
|
Parameters |
Azithromycin |
|
Theoritical Plates |
8335 |
|
Peak Symmetry |
0.9 |
|
%RSD |
0.2 |
The intra- and inter-day variability or precision data are summarized in (Table3). The intra-day precision of the developed LC method was determined by preparing the samples of the same batch. 6. 4 g of sample weighed into six separate 250 mL volumetric flask ,and made upto the mark with diluent . Filtered about 10 mL of above solution through 0.45µm nylon syringe filter by discarding first 4 mL solution. Injected blank, and six replicate injections of repeatability solutions.The %R.S.D,% assay of the assay results was used to evaluate the method precision. The inter-day precision was also determined by the same procedure. The results indicated the good precision of the developed method (Table3).
Table .3
Intra -and inter- day assay precision data(n=12)
|
Sample No. |
Method Precision |
Intermediate Precision |
Over all % RSD (n=12) |
|
1 |
98.3 |
101.8 |
1.0
|
|
2 |
100.2 |
101.3 |
|
|
3 |
100.1 |
98.6 |
|
|
4 |
99.9 |
100.2 |
|
|
5 |
99.9 |
100.3 |
|
|
6 |
100.5 |
101.1 |
|
|
Mean |
99.8 |
100.6 |
|
|
% RSD |
0.8 |
1.1 |
View Within Article Linearity
Linearity was determined for azithromycin in the range of 382–1208 μg/mL. The correlation coefficient (‘r’) value for the drug was >0.999. Typically, the regression equation for the calibration curve was found to be y = 966.6x − 8620.8 for azithromycin.
Robustness
The robustness of an analytical procedure is a measure of its capacity to remain unaffected by small, but deliberate variations in method parameters and provides an indication of its reliability during normal usage.
Robustness of the method was investigated under a variety of conditions including changes of pH of the mobile phase, flow rate, percentage of methanol in the mobile phase and column oven temperature. The standard solution is injected in five replicates and sample solution of 100% concentration is prepared and injected for every condition and % R.S.D. of assay was calculated for each condition. The degree of reproducibility of the results obtained as a result of small deliberate variations in the method parameters has proven that the method is robust (Table 4)
Table 4
Results of robustness study.
|
Robustness parameter |
Level |
% RSD of Results |
|
pH of mobile phase |
8.8 ( Low pH) |
0.1 |
|
9.2 ( High pH) |
0.1 |
|
|
organic composition |
High % organic strength |
0.1 |
|
Low % organic strength |
1.1 |
|
|
flow rate |
1.4 mL/min |
0.2 |
|
1.6 mL/min |
0.1 |
|
|
column oven temperature |
25 |
0.1 |
|
35 |
0.3 |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Conclusion
A simple, specific, linear, precise, accurate and stability indicating RP-HPLC method has been developed and validated for quantitative determination of azithromycin in powder for oral suspension. Statistical analysis proves that method is repeatable and selective for the analysis of azithromycin in powder for oral suspension. As the method seperates the drug from its degradation products it can be employed as a stability indicating one. The developed method can be applicable for pharmaceutical dosage forms and in process testing.
References
[1] N. Kujundzic G. Kobrehel, Z. Banic, Z.Kelneric, B. Koic-Prodic. Azalides (1995) chem Euro J 30:462.
[2] J.E.Kapusnik-under,M.a. sande, H.F.chambers,Farmacos antimicrobianos,in J.Hardman, L.Limbird (Eds.), Goodman&gilman. As Bases Farmacologicas da Terapeutica, ninth ed., Mc Graw Hill, Santiago,1996.
[3] H.G. Fouda, R.P.Schneider (1995) Ter.drug Mon 17:183.
[4] I. Kanfer, M.F. Skinner, R.B. Walker (1998) J Chromatogr A 812:286
[5] J.S. Torano, H.P. Guchelaar, J Chromatogr. B (1998) 720:910.
[6] T. Turcinov, S. Pepeljnjak, J. Pharm.Biom. Anal (1998) 17:97.
[7] R.Gandhi, C.L. Kaul,R. Panchagnula, J.Pharm. Biom. Anal (2000) 23 :1079.
[8] The United States Pharmacopoeia, 24 ed., Rockville: United states Pharmacopeial convention , 2000.
[9] N.Kovacic, J.Marincel, J.Chromatographia (1988) 25:1003.
[10] Validation of analytical methods and procedures :Text and Methodology Q2 (R1):ICH Harmonised Tripartite Guidline. 2007 Nov.

NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






