

 About Authors:
About Authors:
Mr.Sanjay wasudeo upare*, Dr.A.V.Chandewar, Dr.M.D.Kshirsagar, Mr.U.S.Koli and Mr.H.K.Pokale
M-Pharm IV semester (Pharmaceutics department)
Pataldhamal Wadhwani college of Pharmacy,
Yavatmal
*uparesanju81@gmail.com
ABSTRACT:
Lornoxicam is a non steroidal anti-inflammatory drug with analgesic properties. The purpose of this study was to develop a taste masked oral disintegrating tablet of poorly soluble Lornoxicam by direct compression technique with β-cyclodextrin (BCD) complexes using various super disintegrants like sodium starch glycolate, crospovidone and L-Hpc. Prepared tablets were evaluated for different properties like drug content, hardness, friability, disintegration time and in vitro dissolution study. The different formulations showed disintegration time between 21 to 33.66 s. Drug release showed time between the ranges of 5 to 30 min. Among all the formulations, L1 showed 97.79% drug release within 30 min. Thus, L1 was considered best among the other formulations.. The tablets showed enhanced dissolution hence better patient compliance.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1344
INTRODUCTION:
Over a decade, the demand for development of orally disintegrating tablets (ODTs) has enormously increased as it has significant impact on the patient compliance. Orally disintegrating tablets offer an advantage for populations who have difficulty in swallowing. It has been reported that Dysphagia (difficulty inswallowing) is common among all age groups and more specific with pediatric, geriatric population along with institutionalized patients and patients with nausea, vomiting, and motion sickness complications. ODTs with good taste and flavor increase the acceptability of bitter drugs by various groups of population. More than 50% of pharmaceutical are administered orally for several reason and undesirable taste is one of the formulation problem encountered with such oral products.2 Taste of pharmaceutical product is important parameter in governing compliance. Thus taste masking of oral pharmaceutical has become important tool to improve patient compliance and the quality of treatment especially in pediatrics. In inclusion complex formation, the drug molecule fits into the cavity of a complexing agent i.e., the host molecule forming a stable complex. The complexing agent is capable of masking the bitter taste of the drug by either decreasing its oral solubility on ingestion or decreasing the amount of drug particles exposed to taste buds thereby reducing the perception of bitter taste. Vander Waals forces are mainly involved in inclusion complexes. β-cyclodextrin is most widely used complexing agent for inclusion type complexes. It is sweet, nontoxic, cyclic oligosaccharide obtained from starch. Strong bitter taste of carbapentane citrate syrup was reduced to approximately 50% by preparing a 1:1 complex with cyclodextrin. The suppression of bitter taste by cyclodextrin was in increasing order of alpha,gamma, beta cyclodextrin. Lornoxicam is a non-steroidal anti-inflammatory drug (NSAID) of the oxicam class with analgesic, antiinflammatory and antipyretic properties. The mode of action of Lornoxicam is partly based on inhibition of prostaglandin synthesis (inhibition of the cyclooxygenase enzyme). Lornoxicam is absorbed rapidly and almost completely from the gastro-intestinaltract. Lornoxicam is very bitter in taste. Therefore to provide this drug in a more accessible and patient compliant form, in the present study an attempt has been made to mask its bitter taste and formulate it into oral disintegrating tablet.
[adsense:468x15:2204050025]
MATERIALS AND METHODS:
Lornoxicam was gift sample from Luin pharma industries Ltd,Aurangabad. Sodium starch glycolate, crospovidone, L-Hpc, β-cyclodextrin, directly compressible mannitol (Pearlitol SD 200), microcrystalline cellulose and aspartame were obtained as a gift sample from MICRO Labs Pvt. Ltd, Vadodara. All the other chemicals used were of analytical reagent grade.
(i) Preparation of complex of Lornoxicam with β- cyclodextrin
Physical mixture method
The required molar (1:3) quantities of drug and β-cyclodextrin were weighed accurately and mixed together thoroughly in mortar with vigorous trituration for about 3 hrs. These mixtures were then passed through sieve no. 44 and finally were stored in airtight containers till further use.
(ii) Characterization of complex for drug content
Drug content was determined by dissolving 25 mg of complex in suitable quantity of 0.1N HCl and analyzed 1mL of appropriately diluted sample at 376
nm using UV-vis spectrophotometer, Shimadzu 1700
|
Drug : β- Cyclodextrin |
% Drug Content ± S.D |
|
1:3 |
100.48 ± 0.46 |
1. Drug Content Estimation:
Drug content was determined by dissolving 25mg of complex in suitable quantity of 0.1N HCl and analyzed 1ml of appropriately diluted sample at 376nm using UV-vis spectrophotometer, shimadzu 1700
2. Evaluation of Taste of Inclusion complex:
Taste of inclusion complex was checked by time intensity method. For this purpose 10 human volunteers were selected. In this method a sufficient quantity of sample was held in mouth for 10 seconds and volunteers were asked to evaluate the complex for taste. Bitterness levels were recorded immediately. Bitterness values, are based on a 0-3 scale
3 being – strong bitter
2 being – moderate bitter
1 being – slight bitterX being – threshold bitter
0 being – tasteless
These volunteers were instructed not to swallow the granules, which were placed on the tongue. They were instructed to thoroughly gargle their mouth with distilled water after the completion of test.
The results are revealed in Table No.8.1
|
Volunteers |
Bitterness Level after 10 sec |
|
1 |
0 |
|
2 |
0 |
|
3 |
1 |
|
4 |
0 |
|
5 |
1 |
|
6 |
0 |
|
7 |
0 |
|
8 |
0 |
|
9 |
0 |
|
10 |
1 |
FORMULATION OF ORODISPERSIBLE TABLET
Orodispersible tablets were prepared using Lornoxicam and β-cyclodextrin complexes which were prepared by Physical mixture method, variable concentrations of superdisintegrants and other excipients. The powder blends equivalent to the 16 mg of drug were sieved and mixed except for the lubricating agent, mixing of all components and then directly compressed using 8mm concave punches. Tablets were evaluated for various tablet parameters.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Table No.8.2 : Composition of all the formulations ( L1 – L9 )
|
Ingredients |
L1 |
L2 |
L3 |
L4 |
L5 |
L6 |
L7 |
L8 |
L9 |
|
Drug complex eq. to 4mg of drug (1:3) |
16 |
16 |
16 |
16 |
16 |
16 |
16 |
16 |
16 |
|
L-Hpc |
5 |
10 |
15 |
|
|
|
|
|
|
|
Polyplasdone XL 10 |
|
|
|
5 |
10 |
15 |
|
|
|
|
SSG |
|
|
|
|
|
|
5 |
10 |
15 |
|
Mannitol |
78.23 |
73.23 |
68.23 |
78.23 |
73.23 |
68.23 |
78.23 |
73.23 |
68.23 |
|
Mcc |
92 |
92 |
92 |
92 |
92 |
92 |
92 |
92 |
92 |
|
Talc |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
|
Aspartame |
0.5 |
0.5 |
0.5 |
0.5 |
0.5 |
0.5 |
0.5 |
0.5 |
0.5 |
|
Aerosil |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
5 |
|
Magnesium stearate |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
1 |
|
Flavor |
0.27 |
0.27 |
0.27 |
0.27 |
0.27 |
0.27 |
0.27 |
0.27 |
0.27 |
|
Total weight |
200 |
200 |
200 |
200 |
200 |
200 |
200 |
200 |
200 |
Formulation type L1, L2 & L3 contain5%, 10% and 15% Low Substituted hydroxyl propyl cellulose ( L-hpc )as superdisintegrants respectively. Formulation type L4, L5 & L6 contain 5%, 10% and 15% Polyplasdone XL 10 as superdisintegrants respectively. Formulation type L7, L8 & L9 contain 5%, 10% and 15% Sodium starch glycol ate as superdisintegrants respectively.
RESULT AND DISCUSSION
ANALYSIS OF LORNOXICAM
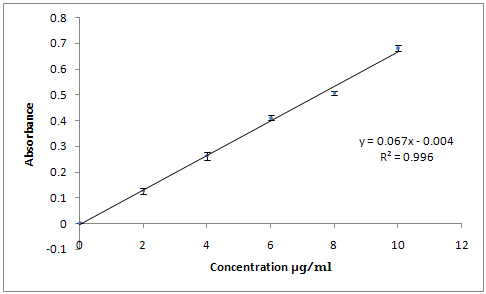
Fig.1 Standard Curve for Lornoxicam
9.2 Excipient Compatibility study.
The possible interaction between the drug and the polymers was studied by FT-IR Spectroscopy.
9.2.1.1 FT-IR Spectroscopy
The possible interaction between the drug and the polymers was studied by FT-IR spectroscopy. The FTIR spectra of pure Lornoxicam and lornoxicam with b- cyclodextrins are as follows.
Fig. No 9.2: FT-IR Spectra of Pure Lornoxicam
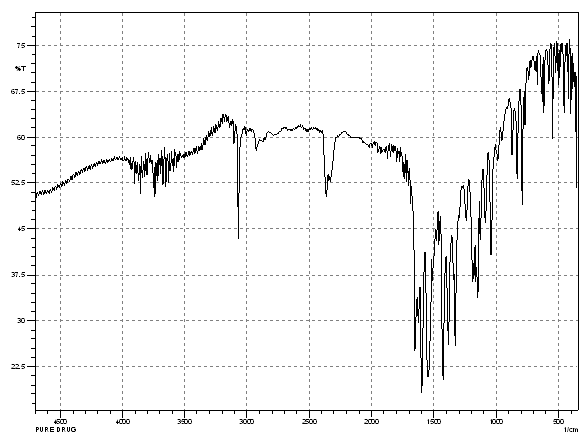
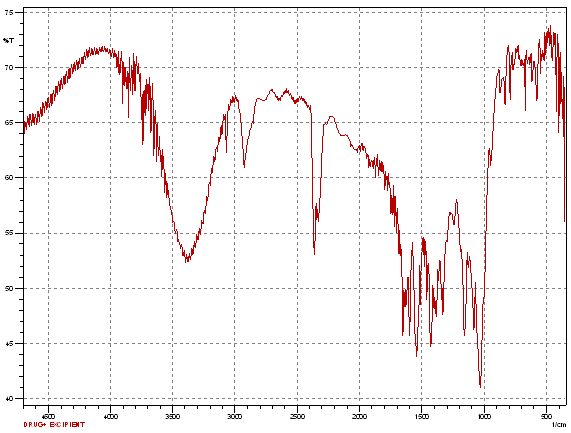
Fig.No.9.3: FT-IR Spectra of Lornoxicam and B- Cyclodextrin
9.2. Micromeritic properties
Preformulation study data of powder blend
In this study, the parameters like bulk density, tapped density, compressibility index, hausner ratio, angle of repose were evaluated. This powder blend altered bulk density values and hence the Carr’s index. The powder blend also improved the flow property
No. 9.2 Blend Characteristics
|
Batch No. |
Angle of Repose (0) |
Bulk Density (gm/cm3) |
Tapped Density (gm/cm3) |
Compressibility Index (%) |
Hausner Ratio |
|
L1 |
19.35 ± 0.23 |
0.293 |
0.331 |
11.48 |
1.13 |
|
L2 |
24.32 ± 0.32 |
0.289 |
0.321 |
12.03 |
1.11 |
|
L3 |
21.06 ± 0.20 |
0.285 |
0.324 |
14.46 |
1.14 |
|
L4 |
21.01 ± 0.26 |
0.280 |
0.317 |
15.23 |
1.13 |
|
L5 |
25.35 ± 0.20 |
0.284 |
0.322 |
13.43 |
1.15 |
|
L6 |
21.19 ± 0.23 |
0.283 |
0.325 |
12.96 |
1.15 |
|
L7 |
23.32 ± 0.23 |
0.284 |
0.323 |
12.03 |
1.13 |
|
L8 |
19.11 ± 0.23 |
0.285 |
0.312 |
14.02 |
1.15 |
|
L9 |
24.35 ± 0.24 |
0.293 |
0.324 |
13.23 |
1.13 |
9.2.2 Angle of Repose
Table No. 9.3 Angle of Repose as an indication of powder Flow Properties
|
Angle of repose (o) |
Types of Flow |
|
Less than 250 |
Excellent |
|
250 - 300 |
Good |
|
300 - 400 |
Passable |
|
More than 400 |
Very Poor |
The angles of repose values given in table 9.3 for all the blends are less than 250 after the calculation. Thus all the blends were having excellent flow properties.
Compressibility Index: Table No.9.4 Relationship between% compressibility and flowability.
|
% Compressibility |
Flowability |
|
5- 15 |
Excellent |
|
12 – 16 |
Good |
|
18 – 21 |
Fair to passable |
|
23 – 35 |
Poor |
|
33 – 38 |
Very poor |
|
› 40 |
Extremely poo |
The values of compressibility index of formulation blends of selected batches given I the table 10 shows that all the values ranged between 5-15%. This implies fairly properties for these blends.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
9.3 Evaluation of Tablet Characteristics
Physicochemical properties of table
A) Thickness
Thickness of tablets were found in the range of 2.93 to 3.07 mm. The thickness of tablet can be calculated with the help of Vernier Caliper.
Table No.9.5Thickness data of the tablets
|
Sr. No. |
Batch Code |
Thickness(mm) + S.D |
|
1 |
L1 |
3.07±0.02 |
|
2 |
L2 |
2.99 ± 0.01 |
|
3 |
L3 |
3.01 ± 0.02 |
|
4 |
L4 |
2.98 ± 0.02 |
|
5 |
L5 |
2.97 ± 0.01 |
|
6 |
L6 |
2.96 ± 0.02 |
|
7 |
L7 |
2.94 ± 0.01 |
|
8 |
L8 |
2.96 ± 0.02 |
|
9 |
L9 |
2.94 ± 0.01 |
B) Hardness, weight variation and friability.
The results of hardness, weight variation and Friability are given in table. The results
showed that weight variation and friability were within the limits.
No.9.6:Hardness, weight variation and friability data of the tablets
|
Sr. No |
Batch Code |
Hardness (Kg/cm2) |
Weight variation(mg) (n=3) |
Friability (%) |
|
1 |
L1 |
3.64 ± 0.12 |
197.63 ± 0.26 |
0.37 |
|
2 |
L2 |
3.58 ± 0.13 |
199.63 ± 0.21 |
0.35 |
|
3 |
L3 |
3.18 ± 0.15 |
198.63 ± 0.26 |
0.38 |
|
4 |
L4 |
3.06 ± 0.17 |
196.63 ± 0.45 |
0.35 |
|
5 |
L5 |
3.24 ± 0.20 |
198.73 ± 0.36 |
0.33 |
|
6 |
L6 |
3.26 ± 0.23 |
199.63 ± 0.26 |
0.31 |
|
7 |
L7 |
3.38 ± 0.17 |
199.63 ± 0.26 |
0.39 |
|
8 |
L8 |
3.08 ± 0.19 |
197.63 ± 0.26 |
0.42 |
|
9 |
L9 |
3.14 ± 0.21 |
197.63 ± 0.31 |
0.36 |
The hardness of all these batches was in the range of 3 – 3.5 Kg/cm2. Such hardness range is enough to give mechanical indicates good Compressibility of blends.
The value of friability for all these formulations was less than the limits of 1.0% which is given in the U.S.P. The friability found in these formulations shows a good strength of tablets to withstand abrasion during transportation and general handling.
C) Drug Content Uniformity
The results for content uniformity are presented in table. The results showed drug content were within the prescribed limits.Table No.9.7: Drug Content uniformity data of the tablets
|
Sr. No |
Batch Code |
Content Uniformity (%) |
|
1 |
L1 |
99.53 ± 0.26 |
|
2 |
L2 |
99.88 ± 0.24 |
|
3 |
L3 |
100.03 ± 0.65 |
|
4 |
L4 |
98.85 ± 0.41 |
|
5 |
L5 |
99.54 ± 0.43 |
|
6 |
L6 |
98.78 ± 0.38 |
|
7 |
L7 |
99.61 ± 0.56 |
|
8 |
L8 |
98.43 ± 0.41 |
|
9 |
L9 |
99.21 ± 0.60 |
Table No.9.8: Tablet wetting time and water absorption ratio of the tablets
|
Sr. No. |
Batch Code |
Disintegration Time(sec)n=3±S.D |
Wetting Time(sec) |
Water absorption ratio |
|
1 |
L1 |
21.00 ± 1.33 |
50.66 ± 1.9 |
65.41 |
|
2 |
L2 |
22.33±2.10 |
47.66 ± 1.5 |
65.31 |
|
3 |
L3 |
26.66±2.50 |
63.33 ± 2.2 |
66.62 |
|
4 |
L4 |
26.00±2.50 |
57.67 ± 2.1 |
67.17 |
|
5 |
L5 |
24.66±2.10 |
54.33 ± 1.9 |
65.31 |
|
6 |
L6 |
28.00±2.20 |
52.66 ± 2.3 |
65.17 |
|
7 |
L7 |
26.66±3.10 |
53.33 ± 2.7 |
66.17 |
|
8 |
L8 |
33.66±2.10 |
65.33 ± 2.6 |
66.98 |
|
9 |
L9 |
32.23±2.20 |
63.00 ± 2.3 |
74.82 |
In Vitro dissolution studies of lornoxicam was carried out in 0.1NHCL for 30 minutes. In order to find out the order of release and the mechanism, which was predominantly influences the drug release from the tablet, the in-vitro dissolution data was subjected to graphical treatment i.e. percentage cumulative drug release Vs time. For In-vitro dissolution study, the prepared batches of Lornoxicam, which were prepared by Direct compression technique using different disintegrating agents like Low-substituted hydroxy propyl cellulose (L-Hpc), Polyplasdone XL10 along with Sodium starch glycolate.
As L-Hpc is preferable in directly compressed tablets. Larger particle size and higher hydroxy propyl content show higher degree of swelling. Polyplasdone XL10 swells rapidly in water. Rapidly disperse in water but does not gel even after prolonged exposure. It has greater rate of swelling compared to other disintegrants and similarly sodium starch gyclolate can act by rapid and extensive swelling. As the superdisintegrants act by swelling and due to swelling pressure exerted in the outer direction or radial direction it causes tablet to burst. The importance of the total surface area in promoting drug dissolution, a water uptake study was performed on lornoxicam orodispersible tablets. Since drug has to dissolved from the interface between drug and water, the maximal water uptake volume can be taken as an estimation of the total surface area available for drug dissolution to take place. Each fiber can act as a hydrophilic channel to facilitate water uptake into the tablet matrix and help increases the total water contact area with drug. In vitro drug release shown by different formulations are,
In L1 formulation, L-Hpc is used as a disintegrating agent in minimum quantity (5%) to obtained the drug release at 30th minute was found to be 97.79%
In L2 formulation, L-Hpc is used as a disintegrating agent with increasing the double quantity i.e.( 10%), the drug release at 30th minute was found to be 85.45%
In L3 formulation, L-Hpc is used up to 15 % and the drug release at 30th minute was found to be 86.75%
In L4 formulation, PolyplasdoneXL10 is used as a disintegrating agent in minimum quantity (5%) to obtained the drug release at 30th minute was found to be 88.05%
In L5 formulation, PolyplasdoneXL10 is used as a disintegrating agent with increasing the double quantity i.e.(10%), the drug release at 30th minute was found to be 86.75%
In L6 formulation, Polyplasdone XL10 is used up to 15 % and the drug release at 30th minute was found to be 89.35%
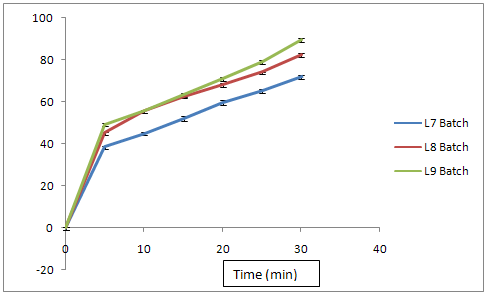
In L7 formulation, Sodium starch Glycol ate is used as a disintegrating agent in minimum quantity ( 5%) to obtained the drug release at 30th minute was found to be 71.80%
In L8 formulation, Sodium starch Glycol ate is used as a disintegrating agent with increasing the double quantity i.e.( 10%), the drug release at 30th minute was found to be 82.20%
In L9 formulation, Sodium starch Glycol ate is used up to 15 % and the drug release at 30th minute was found to be 89.35%
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Table No. 9.9 In-Vitro Drug Release Data of formulation L1 to L9
|
|
Cumulative % Drug Release |
||||||||
|
Time in min. |
L1 |
L2 |
L3 |
L4 |
L5 |
L6 |
L7 |
L8 |
L9 |
|
5 |
55.81 ± (0.21) |
54.47±(0.45) |
53.14±(0.25) |
53.81±(0.1) |
55.14±(0.35) |
55.81±(0.20) |
38.44±(0.25) |
45.12±(0.21) |
49.13±(0.54) |
|
10 |
62.81± (0.17) |
59.49±(0.51) |
60.15±(0.35) |
60.15±(0.35) |
60.15±(0.35) |
61.48±(0.40) |
44.87±(0.59) |
55.50±(0.40) |
55.50±(0.64) |
|
15 |
68.41± (0.55) |
65.10±(0.21) |
68.41±(0.51) |
68.41±(0.50) |
71.05±(0.25) |
69.07±(0.85) |
51.89±(1.0) |
62.46±(0.85) |
63.12±(0.81) |
|
20 |
77.22± (0.75) |
72.62±(0.11) |
74.60±(0.5) |
74.60±(0.3) |
77.22±(0.50) |
75.25±(0.25) |
59.48±(1.1) |
68.02±(0.74) |
70.65±(0.75) |
|
25 |
87.24± (1.0) |
81.36±(0.05) |
80.71±(0.5) |
80.71±(0.80) |
82.02±(0.25) |
82.67±(0.25) |
65.03±(1.10) |
74.17±(0.05) |
78.75±(1.15) |
|
30 |
97.79± (0.11) |
85.45±(0.50) |
86.75±(0.65) |
88.05±(0.75) |
86.75±(0.45) |
89.35±(0.55) |
71.80±(0.7) |
82.20±(1.0) |
89.35±(0.95) |
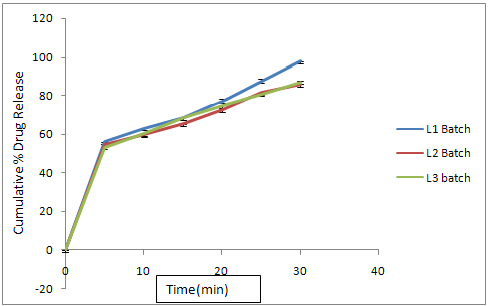
Fig. No. 9.21 % Cumulative Release Profile of Batch L4L5L6
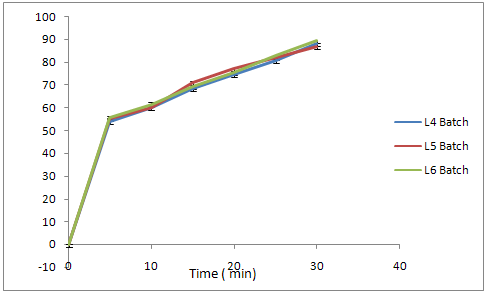
Fig. No. 9.22 % Cumulative Release Profile of Batch L7L8L9
F) Kinetic Release Models:
KINETIC TREATMENT OF DATA OF DISSOLUTION PROFILES OF LORNOXICAM ORODISPERSIBLE TABLETS.
All the findings suggest that the release of drug molecules from the osmotic device has some specific mechanisms. The study of these mechanisms can give an indication of the similarity or differences between the release profiles of various formulations. To study these mechanisms some mathematical equations have been developed by various scientists. These equations are being employed to know the probable mechanism of drug release.Table No. 9.10show the release kinetics of drug for individual polymers.
The release models employed for study of kinetics include zero-order; first-order, Higuchi Eq., the power law and Hixson-Crowell Eq. The exponent of power law determines the probable mechanism by which drug has been released from the osmotic device. The release model was chosen on basis of best fitted criterion. Regression analysis of the equations determines the best fitted model.
The other simplified form of the determination of release mechanism was proposed by Peppas et al. The exponential relation was utilized by these scientists to describe the Fickian and Non-Fickian release behavior of swelling controlled release systems.
The results shown in table no. 9.10the release of maximum batches follows Korsmeyer-Peppas eq., which states that the release of drug might follow the mixed kinetics. It was found that all batches follow Fickian diffusion (n = 0.1 to 0.5).
The results shown in table no. 9.10that the release of six batches follows Peppas eq. and remaining batches follows Matrix eq. This states that the release of drug might follow the mixed kinetics. However all batches follow Fickian diffusion (n = 0.1 to 0.5) as projected by Korsmeyer-Peppas eq. Optimized formulation L1was subjected to curve fitting analysis ,Zero order and first order, Korsmeyer and peppas model optimized formulation L1 fitted best for Matrix.
|
Batches |
bBest fitted model |
Zero order |
1st order |
Matrix |
Peppas
|
Hix-Crow |
Exponent of korsmeyer-Peppas Eq. |
Constant of Korsmeyer-Peppas Eq. |
|||||
|
R |
k |
R |
k |
R |
K |
R |
k |
R |
K |
N |
K |
||
|
L1 |
Matrix |
0.781 |
3.750 |
0.914 |
-0.098 |
0.972 |
18.002 |
0.948 |
-0.002 |
0.935 |
32.043 |
0.303 |
82.5730 |
|
L2 |
Peppas |
0.694 |
3.469 |
0.942 |
-0.067 |
0.960 |
16.779 |
0.885 |
-0.0176 |
0.966 |
34.233 |
0.258 |
21.9231 |
|
L3 |
Peppas |
0.704 |
3.518 |
0.951 |
-0.070 |
0.966 |
17.012 |
0.894 |
-0.018 |
0.989 |
-0.018 |
0.275 |
24.9231 |
|
L4 |
Peppas |
0.708 |
3.537 |
0.954 |
-0.071 |
0.966 |
17.094 |
0.901 |
-0.018 |
0.984 |
33.379 |
0.273 |
91.4677 |
|
L5 |
Peppas |
0.681 |
3.578 |
0.944 |
-0.072 |
0.960 |
17.326 |
0.881 |
-0.018 |
0.983 |
34.730 |
0.283 |
93.1997 |
|
L6 |
Peppas |
0.696 |
3.596 |
0.955 |
-0.074 |
0.962 |
17.390 |
0.901 |
-0.018 |
0.977 |
35.044 |
0.262 |
10.4953 |
|
L7 |
Matrix |
0.791 |
3.164 |
0.955 |
-0.057 |
0.983 |
15.192 |
0.921 |
-0.015 |
0.984 |
24.981 |
0.350 |
92.4677 |
|
L8 |
Peppas |
0.755 |
3.312 |
0.957 |
-0.062 |
0.987 |
26.829 |
0.920 |
-0.016 |
0.987 |
26.829 |
0.322 |
90.1997 |
|
L9 |
Matrix |
0.786 |
3.372 |
0.960 |
-0.065 |
0.981 |
16.195 |
0.931 |
-0.017 |
0.979 |
27.174 |
0.325 |
84.5730 |
Table No.9.10.Kinetic Release Models
10. summary and conclusion
Review of literature indicates that there is a need to improve patient compliance, specially for the bitter drugs like Lornoxicam and hence it was decided to formulate Orodispersible tablet, since it makes possible for the administration of drug without the need of water and by taste masking of drug.
Lornoxicam is the Non-steroidal anti-inflammatory agents, and which is used in the treatment of acute pain conditions. It has also been reported that Lornoxicam has biological half life of 3-5 hrs, so it is desirable to formulate orodispersible tablet which would increase the bioavailability and give rapid onset of action by oral route. It was suggested that orodispersible drug delivery system are easiest approach for technical and logical point of view so, for present study orodispersible drug delivery system was chosen. The drug has very bitter taste; taste plays an important role in patient compliances and acceptability, as far as orodispersible tablets are concerned.
Therefore bitter taste of drug was successfully masked by the use of β-cyclodextrin. The main rational for the formulation of orodispersible tablets are as follows.
• It will be convenient for geriatric, pediatric and mentally ill patients.
• It does not require water at the time of administration.
Lornoxicam is widely used as non-steroidal anti-inflammatory drug. It is very bitter and yet no orodispersible taste masked preparations that might be useful in geriatric, pediatric and mentally ill patients available in the market. Therefore, in the present study an attempt has been made to mask the bitter taste and formulate into a more accessible and patient compliant orodispersible tablets. And to deliver lornoxicam via oral delivery system to give rapid release and onset of action.
Orodispersible tablets of taste masked Drug β-cyclodextrin inclusion complex were prepared by using Mannitol, Mcc, Aspartame, orange flavor, Aerosil, Magnesium stearate and different super disintegrants in varying concentrations. Before preparing the tablets of lornoxicam, the drug and polymer were evaluated for physical properties like bulk density, tapped density and Compressibility index, Hausners ratio. The lornoxicam and polymers showed optimum physical properties.
Nine formulations of Drug β-cyclodextrin inclusion complex (F1-F9) were prepared by varying concentrations of superdisintegrants and Mcc, keeping other excipients constant, these tablets were prepared by direct compression method. Total weight of individual tablet was kept constant i.e.200 mg.
Formulations F1-F3, F4-F6 and F7-F9 were prepared by using L-Hpc, PolyplasdoneXL10 and Sodium Starch Glycolate in 5%, 10%and 15 % concentrations. These tablets were evaluated for: Hardness, Friability, Wt. variation, Thickness, Invitro disintegration time, content uniformity, Disintegration study and dissolution study.
All the formulations showed the satisfactory results and complies with all physical parameters. F1-F9 formulations were found acceptable in Hardness, Invitro disintegration time and dissolution study.
In FTIR spectroscopy, similar peaks were obtained of pure drug and pure drug with excipient, which indicate that there are no any physical or chemical interactions between drug and excipients.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Conclusion:
Nine batches of orodispersible tablets were prepared, by using three types of superdisintegrants with different concentration in all batches.
The following conclusion can be drawn on the basis of the results of the study:
· Bitter taste of Lornoxicam was masked successfully by using beta cyclodextrin using in the ratio of 1:3, which was evaluated by drug content and time sensitivity method.
· Physical properties of drug and polymer were found suitable.
All the tablets formulation showed fair uniformity in drug content (98.43%-100.03%.), and thickness (2.94-3.07 mm ).
· The tablets showed hardness value ranging from 3-3.5 Kg/cm2. In the present study, friability were found to be <1% indicating that friability is within the prescribed limit.
· The disintegration time of all nine formulations was found to have disintegration time in the range 21 – 33.66 sec. L-Hpc having concentration of 5% shows DT of 21 sec, from this it was concluded that, L-Hpc were suitable for use in system to achieve desired invitro disintegration and drug release.
· Wetting time is an important parameter by which determine gain of water to wet the tablet. The L-Hpc found to have wetting time in the range of 47.66 – 63.33 sec.
· In drug release study it was found that L-Hpc showed 97.79% drug release in 30 minutes.
· The kinetic studies revealed that all the formulations followed Korsmeyer-Peppas eq
. Thus, the results of above study clearly indicated that lornoxicam may be formulated as orodispersible tablet using superdisintegrants like L-Hpc, polyplasdone XL10 and sodium starch gyclolate by direct compression method.
12. References:
1) Brahmankar DM, Jaiswal SB, “Biopharmaceutics & Pharmaceutics”, First Edition; pp335(1995).
2) Howard C, Ansel, Nicholas G, Popvich, Loyd V. Allen Jr.; “Pharmaceutical Dosage Forms and Drug Delivery System”; First edition; pp 78 (1995).
3) Jain NK. and Sharma SN; “A Text Book of Professional pharmacy”; Fourth Edition, 6,(1998).
4) Mehta RM; “Pharmaceutics I”; Third Edition;pp 7,238 (2002).
5) Lachman L and Lieberman HA;“Theory and Practice of Industrial Pharmacy”; Third Edition,pp293-294,(1990).
6) Lachman L and Lieberman HA; “Theory and Practice of Industrial Pharmacy”; Third Edition, pp329-335,(1990).
7) Lieberman HA “Pharmaceutical Dosage Forms; Tablets”; Second Edition, Volume I,pp136.
8) Lieberman HA.“Pharmaceutical Dosage Forms; Tablets”; Second Edition,VolumeI,pp 198-1999.
9) Sugharia M. Farumashia, Chem. Pharm. Bull.,30,pp 1396-1400 (1994).
10) Seager H;“Drug Delivery Products and Zydus Fast Dissolving Dosage Forms”, J.Pharma. Tech; 1998,50,pp375-382.
11) Chang RK., Guo X., Burniside BA.and Couch RA. “Fast Dissolving Tablets”,Pharma. Tech.; 2000, 24(6),pp52-58.
12) Dobetti L., “Fast melting Tablets: Development and Technologies”,Pharm. Tech. 2001(suppl.),44.
13) Bhowmik, D. Krishnakanth.,“A Fast Dissolving Tablets”,J.chemical and Pharmaceutical Research, 2009, 1(1): 163-177.
14) Deshmukh VN,; “A Mouth Dissolving Drug Delivery system”, Int. J. Pharmatech Research,2012,Volume.No.4,pp 412-421.
15) Chaturvedi AK. and Verma A.,” Fast Disintegrating Tablet Technology”, Int. J. Pharmaceutical Science and Research,2011, Volume. 2,(12)pp 3046-3050.
16) Seager H., “Drug Delivery Products and zydus Fast Dissolving Dosage Forms”, J. Pharm. Pharmacology,; 1998,50,pp375-382.
17) Gupta AK., Mittal A., “Fast Dissolving Tablet”, A Pharma Innovation, 2012,Volume 1.
18) Chatap VK.,“A Review on Taste masking Methods on Bitter Drugs”, pharmainfo.net, 2007, Volume 5.
19) Sampath K. Bhowmik D.., “Recent Trend in Ion Exchange Resins Used in Pharmaceutical Formulation, The Pharma Research, 2010,4;138-148.
20) Behera S., Panda C.“A Review on Fast Dissolving Tablet”, webmed central Pharmaceutical Sciences, 2010;1(11).
21) Martin GL.,“Ion Exchange and Adsorption Agents in Medicines” Little Brown, Boston 1955.
22) Jain NK.; Advances in controlled and Novel drug Delivery; First Edition.pp 290 (2001).
23) Ion Exchange Resins and sustained release; Swarbik, J.; Encyclopedia of pharmaceutical Technology, volume-8,pp203-217.
24) Indian Pharmacopoeia, Vol.-I,pp494(1996).
25) www. DrugBank.Com
26) www. Wikipidia.com
27) Paul J., Marian E., “Handbook of drug Excipient” Sixth Edition.
27) Ghosh A.; Biswas S:;“Preparation and Evaluation Silymarin β-Cyclodextrin Molecular Inclusion Complexes.” J Young Pharm. 2011 Jul-Sep; 3(3): 205–210.
28) Patel SJ. Sheth, SK. “Formulation and Evaluation of Taste masked oral disintegrating tablet of Lornoxicam.” Int. J. of Pharma and Bioscience. 2010.
29) Chandrasekhar Y.; Ramya RR.; “Orodispersible Tablet of Lornoxicam with Natural and Synthetic Superdisintegrants.” Int. J. of Pharmacy and Technology. 2011.
30) Yunixia, B., Sunada, H., Yonezawa, Y., and Danjo, K.; “Evaluation of rapidly disintegrating tablet prepared by direct compression method”,Drug. Dev. Ind.Pharm., , 25(5), PP 571-581, ( 1999).
31) Remington, The science and practice of pharmacy; 19 th Edition, volume II.
32) Indian pharmacopiea, vol-I pp 156 (1996).
33) Indian pharmacopiea, vol-I pp226 (1996).
34) British pharmacopiea, vol-I pp511 (1996).
35) Raymond .c. Rowe, Paul J Sheskey And Paul J Weller, “Handbook Of Pharmaceutical Excipients”, fourth edn, pp 373-377.
36) Vijaya K G and Mishra D N, “rapidly disintegrating oral tablets of meloxicam, Indian Drugs 43(2), pp 117-121.
37) G D Gupta, And R S Gaud, “ Formulation And Evaluation Of Nimuselide Dispersible Tablet Using Natural Disintegrants, “Indian journal of pharmaceutical sciences,”pp339-342, (sept-oct 2000)
38) Willium r pfister and Tapash K Ghosh, “ Orally Disintegrating Tablets Products, Technologies And Development Issues, Pharmaceutical Technology, pp 142, (Oct-2005).
39)Larry L. Augsburger; and Mark J. Zellhofer, “Tablet Formulation,” Encyclopedia Of Pharmaceutical Technology, (Jan, 2002).
40) Wilson CG, Washington N, Peach J, Murray GR, Kennerley J.The behavior of a fast dissolving dosage form (Expidet) followed by g-scintigraphy.Int J Pharm. 1987; 40: 119–123.doi:10.1016/0378-5173(87)90056-1
41) Fix JA.Advances in quick-dissolving tablets technology employing Wowtab.Paper Presented at: IIR Conference on Drug Delivery Systems. 1998 Oct.; Washington DC, USA.
42) Virely P, Yarwood R. Zydis – a novel, fast dissolving dosage form. Manuf Chem. 1990; 61: 36–37. 43) Jaccard TT, Leyder J. Une nouvelle forme galenique: le lyoc.[A new galenic form: lyoc] Ann Pharm Fr. 1985;
43: 123–131.PMid:4091454
44) Parakh SR, Gothoskar AV. A review of mouth dissolving tablet technologies.Pharm Technol. 2003; 27: 92-100.
45) Kuchekar B S, Atul C Badhan, Mahajan HS. Mouth dissolving tablets: a novel drug delivery system.
46)Raymond C Rowe, Paul j sheskey, Paul j Weller (2003). ”Handbook of pharmaceutical excipients”, fourth edition.
47)Masaki K (1995). Intrabuccally disintegrating preparation and production thereof. US Patent 5,466,464.
48)Pebley WS, Jager NE, Thompson SJ (1994). Rapidly disintegrating tablet.US Patent 5, 298, and 261.
49)Ringard J, Guyot-Hermann AM (1988). Calculation of disintegrant critical concentration in order to optimize tablets disintegration. Drug Dev Ind Pharm.14: 2321–2339.
50)Watanabe Y (1995). New compressed tablet rapidly disintegrating in the mouth using crystalline cellulose and a disintegrant. Biol Pharm Bulletin. 18: 1308–1310.
51)Wehling F, Shuehle S (1996). Base-coated acid particles and effervescent formulation incorporating same. US Patent 5,503,846.
52)Seager H (1998). Drug delivery products and the Zydis fast dissolving dosage form. J Pharm Pharmacol. 50: 375–382. 53)Wehling F, Shuehle S, Madamala N (1993). Effervescent dosage form with microparticles.US Patent 5,178,878.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






