

{ DOWNLOAD AS PDF }
 ABOUT AUHTORS
ABOUT AUHTORS
Jaha Sultana Mohammed
Pelcat Formulation PVT
sohnivya786@gmail.com
ABSTRACT
The purpose of cleaning validation is to establish the documented evidence with high degree of assurance that the cleaning process followed as per standard operating procedure for cleaning the equipment used for the processing, consistently and concurrently yields the results not exceeding predetermined acceptance limit. The main objective of this particular study is to develop some understanding for the process of validation and its type along with importance of cleaning validation in pharmaceutical industry to prevent cross contamination. This topic includes Types of validation, cleaning validation, Levels of cleaning Validation, Cleaning mechanisms, cleaning agents used and process followed by pharmaceutical industry to achieve cleaning validation. The various methods used for cleaning validation are clearly discussed in this review.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-2460
|
PharmaTutor (ISSN: 2347 - 7881) Volume 5, Issue 1 Received On: 27/08/2016; Accepted On: 04/10/2016; Published On: 01/01/2017 How to cite this article: Mohammed JS; Validation in Pharmaceutical Industry : Cleaning Validation - A Brief; PharmaTutor; 2017; 5(1); 8-18 |
VALIDATION
INTRODUCTION
It is known today, because there is need to maintain quality, consistency and above all public safety. Validation is a rapid growing and evolving subject. Over a past 15 years, machine automation and process control in the pharmaceutical industry has caused additional concerns relating the validation of the processing systems. Validation is responsible for providing higher degree of assurance for the product. The foundation of validation, the methodology behind validation, and the need for validation will likely remain a key aspect of the industry we work in [1, 2].
DEFINITIONS OF VALIDATION
According to FDA (FOOD AND DRUG ADMINISTRATION) VALIDATION is a procedure for production and process control designed to assure that the drug products have their identity, strength, quality and purity. According to FDA guidelines in May 1987, the validation package must provide the necessary information and test procedures required to prove that the system and the process meet the specified requirements. The qualification is done in three ways
- Installation Qualification (IQ)
- Operational Qualification (OQ)
- Process Qualification (PQ)
- INSTALLATION QUALIFICATION: It is a process of verification that the equipment/ system is installed in a proper manner and that all of the devices are placed in an environment suitable for their intended range of use.
- OPERATIONAL QUALIFICATION: It is a process of verification that equipment performs as expected throughout the intended range of use.
- PROCESS QUALIFICATION: It is a process of verification that the system is repeatable and consistently producing a quality product [1].
WHY VALIDATION IS NEEDED?
- To makes working easier and safer.
- To sets the recognized level of Quality.
- To facilities integrability.
- To enhances efficiency.
- Validation Streamlines production, increases productivity [1].
WHEN DOES VALIDATION BEGIN?
Ideally validation starts in the very beginning, in the laboratory. In the lab, scientists discover exactly how the product reacts, as well as the parameters that are required to produce such a product. They learn under what conditions the product fails or becomes unstable, unusable and when its quality begins to suffer. Once the laboratory has established the boundary processing criteria, this information can then be used for establishing requirements for validation. Validation begins with good up-front definition of the validation process. Typically, information is structured so that the functional requirement [1].
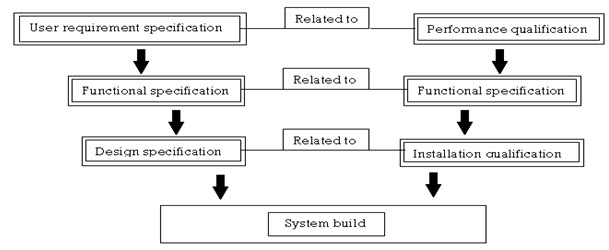
DESCRIPTION
- Operational tests that encompass the performance of the overall system should be designed to guarantee the end pharmaceutical product meets or exceeds the designed intent of the drug.
- The functional specification describes the detailed operation of the equipment, from which an operational qualification test plan can be constructed.
- The design specification usually consists of the electrical schematics, part description and the details required constructing the system. This information usually includes the installation qualification criteria required to adequately insure that the machine is being installed in an environmental suitable for its use that means proper location, proper voltage, classification.
- This total information helps to qualify the system and the processes.
WHEN DOES VALIDATION ENDS?
Validation of a system never truly ends. Once a new system and process have been validated the system still requires maintenance, periodic calibrations and adjustment. Therefore, the process is always under scrutiny and constant evaluation [1].
DEPARTMENTS RESPONSIBLE:
- Site validation committee (SVC): Develop Site master Validation plan, Prepare/execute/approve validation Studies
- Manufacturing department: Prepares the batches as a routine Production batch
- Quality assurance: Ensure compliance, see that documentations/procedures are in place, approves protocols and reports
- Quality control: Perform testing and reviews protocol and report as needed [3].
RESPONSIBLE AUTHORITIES FOR VALIDATION:The validation working party is convened to define progress, coordinate and ultimately, approve the entire effort, including all of the documentation generated. The working party would usually include the following staff members, preferably those with a good insight into the company's operation.
- Head of quality assurance
- Head of engineering
- Validation manager
- Production manager
- Specialist validation discipline: all areas [3]
|
Department /Designation |
Responsibility |
|
Manager Production |
Responsible for manufacturing of batches and review of protocol and report. |
|
Manager QC |
Responsible for analysis of samples collected |
|
Executive QC |
Responsible for samples collection and submission to QC |
|
Manager Maintenance |
Providing utilities and engineering support |
|
Executive Production |
Responsible for preparation of protocol and manufacturing of validation batches |
|
Manager QA |
Responsible for protocol authorization and preparation of summary report. |
- Analytical Method Validation
- Process Validation
- Software Validation
- Cleaning validation
AnalyticalMethod Validation:
Confirmation by means of examination and provision of object evidence that the particular requirements for a specific intended use can be consistently fulfilled.
Process Validation:
Establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its predetermined specifications and quality attributes.
SOFTWARE VALIDATION:
When software or automated data processing systems are used as a part of the production or the quality system, the manufacturer shall validate computer software for its intended use according to an established protocol. All software changes shall be validated before approval and issuance
CLEANING VALIDATION:
Cleaning validation is the process of assuring that cleaning procedures effectively remove the residue from Manufacturing equipment / facilities below a predetermined level [1, 4].
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
CLEANING VALIDATION
INTRODUCTION
Cleaning validation is a documented process that proves the effectiveness and consistency in cleaning of pharmaceutical equipment. It is necessary to have effective cleaning programs in place because of the regulatory and compliance requirements. There is however a more fundamental reason and that are moral requirements to produce products that are as pure and free from contamination to the extent that is possible and feasible. Cleaning validation programmers are necessary simply to prevent our manufactured products from being contaminated. There are two types of contamination
- Cross contamination of one product into another
- Product contamination by a foreign material
Cross contamination is usually thought of in terms of an active ingredient from one product carrying over into a subsequently manufactured product. The second type of contamination is by foreign particles these may be bacterial in nature or could represent parts of the equipment such as gasket or linings [1].
DEFINITION
Cleaning Validation is the process of providing documented evidence that the cleaning methods employed within a facility consistently controls potential carryover of product (including intermediates and impurities), cleaning agents and extraneous material and also confirms a reliable cleaning procedure [1].
OBJECTIVES:
- Increases equipment utilization.
- Extension of equipment life
- Provide assurance of cleaning
- Provide assurance of product purity, safety, efficacy and quality
- Prevention of product cross contamination by byproduct, residual product etc.
- Solvent reduction
- Shorter cleaning time
- Work safety
- Cost effectiveness [5].
WHY CLEANING VALIDATION?
- To verify the effectiveness of cleaning procedures and to ensure no risks are associated with cross contamination of active ingredients or detergent/sanitizer.
- To protect product integrity and to avoid cross contamination for internal control and compliance.
- To reuse the equipments and make the process more cost effective.
- To combat the regulatory authorities (FDA, USFDA, cGMP).
- It is a regulatory requirement in Active Pharmaceutical Ingredient product manufacture
- To increase Equipment Utilization [4].
WHEN CLEANING VALIDATION?
The cleaning validation is done in following case
- Initial qualification of a process/equipment
- Critical change in a cleaning procedure and formulation
- Significant change in equipment
- Change in a cleaning process and cleaning agent.
LEVELS OF CLEANING:
Levels of cleaning mainly depend on the following aspects:
- The equipment usage
- The stage of manufacture
- The nature of the potential contamination
- Level 0 cleaning (Batch to Batch):
It is an in- campaign batch to batch change over requiring no validation. It can be of two types:
- Done between intermediate steps in the same manufacturing process.
E.g. Step B is performed immediately after Step A for the same product line.
- Done between the steps in the manufacturing processes of 2 batches of the same product.
E.g. For given equipment, Step A of the first batch is to be followed by the manufacturing of Step A of second batch.
- Level 1 cleaning (Product to Product):
It is performed when cleaning after an intermediate or final product step of one product followed by the production of an intermediate step of the other product.
E.g. For a given equipment, Step A of drug A followed by step A of drug B.
- Level 2 cleaning (Product to Product):
It is performed when cleaning after an intermediate or final product step of one product followed by the production of final step of the other product [6, 7].
E.g. Step A of drug A followed by last step of drug B.
PRINCIPLE
- Cleaning procedures must strictly follow carefully established and validated methods of execution. This applies equally to the manufacture of pharmaceutical products and active pharmaceutical ingredients (APIs). The objective of the Cleaning Validation is the confirmation of a Reliable cleaning procedure so that the analytical monitoring may be omitted or reduced to a minimum in the routine phase [7].
DOCUMENTATION
A Cleaning Validation Protocol is required laying down the procedure on how the cleaning process will be validated. It should include the following:
- Responsibilities for performing and approving the validation study;
- Description of the equipment to be used;
- The interval between the end of production and the beginning of the cleaning procedures;
- Cleaning procedures to be used for each product, each manufacturing system or each piece of equipment;
- The number of cleaning cycles to be performed consecutively;
- Any routine monitoring equipment;
- Sampling procedures, including the rationale for why a certain sampling method is used;
- Clearly defined sampling locations;
- Data on recovery studies where appropriate;
- Analytical methods including the limit of detection and the limit of quantization of those methods;
- The acceptance criteria, including the rationale for setting the specific limits; Other products, processes, and equipment for which the planned validation is valid according to the “bracketing” concept; and
- When Re-validation will be required.
The Cleaning Validation Protocol should be formally approved by the Plant Management, to ensure that aspects relating to the work defined in the protocol, for example personnel resources, are known and accepted by the management. Quality Assurance should be involved in the approval of protocols and reports [6].
CLEANING MECHANISMS
Cleaning involves removing an unwanted substance (the contaminant) from a surface (the equipment to be cleaned). The chemistry of cleaning includes several mechanisms that serve to remove or assist in removing the contaminants from the equipment surfaces. Understanding (or at least being aware of) cleaning mechanisms can assist in the selection of the proper cleaning agent; more importantly, it can assist in the proper design of the overall cleaning process [5]. It includes following mechanisms: Solubility, Solubilization, Emulsification, Dispersion, Wetting, Hydrolysis, Oxidation, Physical removal, Antimicrobial action.
CLEANING AGENTS USED IN CLEANING VALIDATION:
- These are used in pre wash and rinsing stages of the operation.
- Properties:
- Compatibility with equipment surface.
- Ability to solubilize the residues within a reasonable contact time.
- Anti microbial properties.
- Frequency of use and employee safety considerations.
- Has a mode and method of application.
E.g.:- automatic/manual cleaning, pH, temperature concentration requirements.
- They include materials such as: Organic Solvents like ethanol, acetone etc, Aqueous Cleaning, Water, Surfactants, Non ionic Detergents, Chelating agents, Acids and Bases, Oxidants like hydrogen peroxide Para acetic acid etc., Antiseptics [1, 5].
CLEANING VALIDATION PROCESS:
Cleaning validation process involves eight stages that are shown below:
Stage-1: Establishment of acceptance criteria
Stage-2: Cleaning procedure
- Identification of the equipment
- Characterization of the products (Previous: activity/toxicity, solubility, subsequent: dosage, lot size)
- Determination and characterization of the cleaning agents
Stage-3: Sampling Procedure and necessary validation of samples
- Swab method
- Rinse method
- Coupon method
- Solvent method
- Placebo method
Stage-4: Analytical method and its validation
Stage-5: evaluating equipment surface
- Worse case location to be identified
- Volume and type of rinsing solvent used
- Equipment surface area
Stage-6: Validation protocol
Stage-7: Validation report
Stage-8: revalidation
Cleaning validation process is explained by following diagram [6].

Stage-1: Acceptance criteria:
The Cleaning Validation should demonstrate that the procedure consists removes residues of the substance previously manufactured down to levels that are acceptable and that the cleaning procedure itself does not contribute unacceptable levels of residual materials to the equipment. The limits set should be practical, achievable and justifiable.
- Limiting the level based on toxicity data:
An Acceptable Daily Intake (ADI) is calculated with suitable safety factors applied and this is converted to the maximum allowable carryover to the API [8].
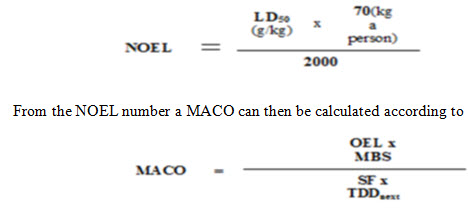
Where,
MACO: Maximum Allowable Carry over: acceptable transferred amount from the investigated product (“previous”).
NOEL: No Observed Effect Level
LD50: Lethal Dose 50 in g/kg animal. The identification of the animal (mouse, rat etc.,) and the way of entry (IV, oral etc., ) is important.
2000: 2000 is an empirical constant
TDDnext: Largest normal daily dose for the next product
MBS: Minimum batch size for the next products (where MACO can end up)
SF: Safety factor
- Pharmacological Dose Method:
The philosophy is to reduce the levels of residual product in each pieces of equipment, such that no greater than 1/1000 of the normal therapeutic dose will be present per typical dose of the next product to be run in the equipment. The validation protocol should include a calculation, which ties this philosophy to the acceptance criteria for the samples to be tested [6].
MAXIMUM ALLOWABLE CARRY OVER (MACO)
MAC = I/J x K/L x M
Where,
I = smallest strength of product ‘A' (previous product) manufactured (safety factor 1000).
J= maxim number of dose units of product ‘B’ (next product) administrated / day.
K= number of dose units per batch of product ‘B’.
L= equipment surface area in common between products ‘A’ and ‘B’ expressed as square centimeters.
M= sampling surface area
(Product ‘A’ is previous product &product ‘B’ is next product).
- Limiting the level of product which could appear in the following Products:
Limits from 10ppm up to 0.1% (based on the ICH impurity document which indicates that up to 0.1% of an individual unknown or 0.5% total unknowns material may be present in the product being tested ) Note FDA Statement on 0.1% impurities.
It is also necessary to evaluate the ability of the cleaning procedure to remove any cleaning agents introduced. The acceptance criteria for the residual-cleaning agents should reflect the absence of these materials, within the range of the capabilities of the assay and sampling methods. The individual company must decide on the Acceptance Criteria which are justifiable for their particular situation [7, 8].
Stage-2: Cleaning procedures:
Written cleaning procedures for each piece of equipment and process1 must be prepared. It is vital that the equipment design is evaluated in detail in conjunction with the product residues to be removed, the available cleaning agents and cleaning techniques when determining the optimum cleaning procedure for the equipment. If one cleaning procedure has been shown to be adequate for a number of products, then it is only necessary to have one cleaning SOP for those products for each piece of equipment. Cleaning procedures should be sufficiently detailed to remove the possibility of any inconsistencies during the cleaning process [7, 8].
- Equipment parameters to be evaluated:
- Identification of the equipment to be cleaned
- Difficult to clean areas
- Property of materials
- Ease of disassembly
- Fixed or not
- Residues to be cleaned
- Cleaning limits
- Solubility's of the residues
- Length of campaigns
- Cleaning agent parameters to be evaluated
- Preferably materials that are normally used in the process
- Detergents available (as a general guide, minimize use of detergents unless absolutely required)
- Solubility properties
- Environmental considerations.
- Health and safety considerations
- Cleaning techniques to be evaluated
- Manual cleaning
- CIP (Clean-in place)
- COP (clean-out-of-place)
- Semi automatic
- Automatic
- Time considerations
- Number of cleaning cycles
- Other requirements:
Procedures must be determined to be operator independent i.e. rugged and reproducible, during the validation studies .The cleaning documentation should include the following items in order to ensure that it can be followed reproducibly and maintained subsequent to Validation [8].
- Detailed definition of levels of cleaning to be performed.
- Detailed description of cleaning methods.
- The necessity to inspect and verify equipment cleanliness prior to manufacture of next batch should be stated in the SOP and recorded on the batch record.
- The SOP should detail where verification of cycle parameters (if automated) and checklists (for complex manual procedures) is necessary.
- Where microbial contamination may be an issue, consideration should be given to the integrity of the vessel prior to manufacture.
Written cleaning procedures may also include additional items not specified above, these would include, as an example, the steps needed to protect the equipment from Contamination after cleaning.
Stage-3: Sampling:
The two methods of sampling generally employed are swab and / or rinse sampling. (If neither or these methods are shown be a scientifically sound method for testing in a specific instance then an alternative is to consider testing the next product.) The selection of either of these techniques must be consistent with sound scientific judgment and must support the objective of the study, which is to demonstrate that the amount of residual material in the equipment has been reduced to acceptable levels [6].
Each method is described in brief below.
- Swab:
Swab sampling does not cover the entire equipment surface area therefore sites must be chosen with care. It is important that, as a minimum, the swab sites represents worst case locations on the equipment and that the result is then extrapolated to account for the total product contact surface area. This calculation makes it possible to make a worst case determination of potential carryover into subsequent product.
- Due to the nature of this method which employs physical forces as well as chemical forces it may be necessary to perform sampling technique evaluation.
- Swabbing efficiency (% recovery) for the swabbing method must be determined.
- It is necessary to ensure that extractable of the swab do not interfere with the sampling method.
- Using this technique it is possible to sample insoluble residues due to the physical action associated it.

Swab accuracy determines a method’s ability to recover the compound of interest directly from the swab head.
There are a variety of swabs to pick from, but when a change in swab type takes place, swab specificity also needs to be revalidated.
Limitations:
- An invasive technique that may introduce fibers.
- Results may be technique dependent.
- Swab material and design may inhibit recovery and specificity of the method.
- Evaluation of large, complex and hard-to reach areas difficult (e.g. crevices, pipes, valves, large vessels).
- Rinse:
In obtaining rinse samples, location, timing and volume are important considerations
- The solvent rinse occurs after cleaning has been completed
- This method is not as direct as swabbing but will cover the entire surface.
- This method allows much greater ease of sampling than swabbing
- A reduced no of samples are required to generate a carryover figure.
Limitations:
- Residues may not be homogeneously distributed.
- Inability to detect location of residues.
- Rinse volume is critical to ensure accurate interpretation of results
- Coupon sampling:
Coupons of the same materials of construction as the item to be cleaned can be affixed to the equipment, spiked with the product, subject to the cleaning procedures and then submitted to the laboratory for direct analysis and recovery studies.
Advantages:
- Allows for direct surface sampling.
- Useful in cleaning method development.
- Reduced variability in recovery.
- Useful in evaluation of equipment materials of construction.
Limitations:
Coupon may not be representative of equipment contamination or cleaning as it is separate from primarily surface.
- Invasive
- Might interfere with the cleaning process.
- Solvent Sampling:
This technique uses a solvent not normally employed in the cleaning process to maximize recovery residues.
Advantages:
- Commonly used in bulk chemical facilities
- Applicable for actives, cleaning agents, Excipients
- Less technique dependent than swabs.
- Usually affords more analytical specificity, less recovery loss than swabs.
- Allows sampling of a larger surface area.
- Allows sampling of porous and delicate surface
- Maximizes recovery to rinse.
Limitations:
- May require operator protection and other safety and environmental protection measures.
- May require more than one sampling for broad spectrum analysis.
- Reduced physical sampling of the surface.
- May be difficult to accurately define the controlled area sampled, therefore usually used for rinsing an entire piece of equipment such as a vessel.
- May require the removal of solvent prior to equipment use for production [8].
- Placebo and Product Sampling:
Placebo sampling can be used to detect residues on equipment thorough the processing of a place to batch subsequent to the cleaning process. Product sampling is similar to placebo sampling except that it uses actual product.
Advantages
- Points of product contact identical for the two batches
- Applicable for hard to reach surfaces.
- Require no additional sampling steps.
Limitations
- Difficult to determine recovery
- Lowers analytical specificity and inhibits detectability
- Residues may not be homogenously distributed.
- No direct measurement of residues on product contact surfaces.
Stage-4: Analytical methods:
In order for the analytical testing of the cleaning validation samples (swabs or rinses) to yield meaningful results, the analytical methods used should be validated. This should be documented. The basic requirements are:
- The ability to detect the target substance(s) at levels consistent with the acceptance criteria
- The ability to detect the target substance(s) in the presence of other materials that may also be present in the sample (selectivity)
- The analytical method should include a calculation to convert the amount of residue detected in the sample to 100% if the recovery data generated indicates a recovery outside of an allowed range.
The analytical methods generally employed in cleaning validation are listed below:
- UV visible spectrophotometer
- pH measurement
- Direct surface analysis
- Conductivity measurement
- Titration
- High performance liquid chromatography
- Thin layer chromatography
- Capillary zone electrophoresis
- Fourier transform infrared
- Enzyme linked immune sorbent assay
- Atomic absorption/ ion chromatography [5, 6].
Stage-5: Evaluating equipment surfaces:
- Worst case location to be identified:
- Solubility: less soluble products more worse the situation
- Potency: more potent product more worse the situation
- Toxicity: more toxicity more worse the situation
- Strength of product: lowest strength of previous product more worse the situation
- Maximum daily dose of next product: highest the number of daily doses more worse the situation
- Common equipment surface area: largest common surface area of equipment more worse the situation
- Volume and type of rinsing solvent to be used:
- Various type of rinsing solvents are used such as alcohol, methanol , some organic acids (acids) , hydrogen peroxides
- The volume of rinsing solvent used is approximately 15-20ml but the selection of volume is based on type of sampling procedure and analytical method used
- Equipment surface area:
The surface area of the cleaning equipment or any substances should be calculated by using relevant formulas of surface area. The calculation of surface area is based on length, width, size and shape [7, 8]
Stage-6: Validation protocols:
A Validation Protocol is necessary to define the specific items and activities that will constitute a cleaning validation study. The protocol must be prepared prior to the initiation of the study and must either include or reference the documentation required to provide the following information:
- The objective of the study:
- What cleaning process is to be validated (indicating the product to be removed and the equipment from which it is to be removed)?
- If this study is to be employed to demonstrate the acceptability of the cleaning procedure for a group of products the rational for doing so should also be detailed here
- The cleaning procedure(s) to be validated should be identified i.e. cleaning agents, soakage times, equipment parameters, number of cleaning cycles etc.
- Scope of the study:
- The company must evaluate the process and determine which residues are to be tested for and which are not to be based on sound scientific rational.
- What residues (including cleaning agents) are to be tested for, why those residues how many times the study should be run before a report is compiled and recommendations made.
- Listing of the process parameters to be verified:
- This is particularly necessary when automated or semi-automated cleaning techniques are to be employed.
- Sampling and inspection procedure to be used:
- The types of sampling methods to be used, where the samples are to be removed from and how many samples are to be taken. Any particular requirements should also be stated i.e. for sterile sampling / sampling light sensitive products.
- An equipment sampling diagram should be referenced.
- Personnel responsibilities during the study
- Test methods to be used [7, 8].
Stage-7: Validation reports:
A validation report is necessary to present the results and conclusions and secure approval of the study. The report should include the following:
- Summary of the procedures used to clean, sample and test
- Physical and analytical test results or references for same, as well as any pertinent observations
- Conclusions regarding the acceptability of the results, and the status of the procedure(s) being validated
- Any recommendations based on the results or relevant information obtained during the study including revalidation practices if applicable.
- Approval of conclusions
- Review any deviations for the protocol that occurred.
Stage-8: Revalidation:
Periodic review and revalidation are methods by which the performance of a validated cleaning process is evaluated to ensure that a state of control is maintained. The rationale for the revalidation frequencies should be appropriately documented and justified. Changes which should require evaluation and likely re-validation include but not limited to
- Changes in the cleaning procedure.
- Changes in the raw material sources.
- Changes in the formulation and/or process of products.
- New products.
- Changes in the formulation of detergents.
- New detergents.
- Modifications of equipments.
CONCLUSION
In the present work, an industrial has taken some procedure regarding to cleaning. The procedure was identified to be validated as cleaning validation. All the equipments were selected from cross contamination point of view based on the matrix approach. From this study, it may be concluded that cleaning validation is an important aspect in assuring the high degree of assurance to the product quality. It plays vital role in assuring and preventing cross contamination and also provides the assurance for the effectiveness of cleaning method chosen. Not only this we can also observe the results of cleaning validation are within the acceptance limit or not and if it is within limit hence the objective of the company to have an effective cleaning program is well documented and ultimately the results were achieved. If not within limit again revalidation takes place.
REFERENCES
- Kathirsan; A text book of methods of validation by, validation page no: 1- 6, cleaning validation page no: 47-72.
- Anjaneyulu, Marayya; A text book of Quality assurance and quality management in pharmaceutical industry; Good manufacturing practice page no: 144-145.
- Phviral: Process validation. An essential process in pharmaceutical industry Pharmainfo.net, 2010-01-10.
- Monohar A.Potdar; A text book of current good manufacturing practice; pharmaceutical validation page no: 413-462.
- Destin A.Leblanc; A text book of validated cleaning technologies for pharmaceutical manufacturing.
- Active pharmaceutical ingredient committee: cleaning validation in active pharmaceutical ingredients manufacturing plant, aic.cefic.org/pub/4cleaningvalidation9909.pdf, September 1991.
- Health sciences authority: regulatory guidance, cleaning validation, has.gov/publish/…/guide-moa-008-006-web.pdf, December 2008.
- Cleaning Validation for Biopharmaceutical Manufacturing at Genentech, Inc. Part 2, pharmaceuticalvalidation.blogspot.in






