

{ DOWNLOAD AS PDF }
ABOUT AUTHORS:
Jitendra Kumar Badjatya
Deputy Manager-DRA,
Montajat Pharmaceutical Company Limited, Dammam, KSA
jeetbadjatya@gmail.com
ABSTRACT
A Generic Product must meet the standards established by China Food and Drug Administration (CFDA) to be approved for marketing in China respectively. This study covers the introduction to generic drugs, in China regulatory authorities. It also includes the requirements and registration of Generic Drugs.
Many large pharmaceutical companies have increased their presence in emerging markets in recent years like China markets is predicted to be the second largest market after US in Pharmaceuticals. China is moving past its phase as a supply market for ingredients and generic finished drugs. So, is more important to know about Regulatory considerations and Registration process of Generic Drugs. This article focuses in particular on pharmaceuticals companies, the Registration process of Generic Drugs, and their activities in the China.
INTRODUCTION
Regulatory affairs
The pharmaceutical industry is one of the most regulated industries. No drug would be available in the market until and unless it get approved by Regulatory Authorities. Regulatory Affairs is a specialized profession in the pharmaceutical sector. Regulatory Affairs oversees with laws and regulations pertaining to the development, marketing and of drug products. Regulatory Affairs acts as point of contact between the company, its products and regulatory authorities with respect to registration.[1]
Regulatory Affairs interacts with International and domestic drug regulators [e.g., MHLW (Japan), CFDA (China), EMA (Europe), FDA (United States), CDSCO (India) etc] to assure:
Pharmaceutical Markets
Even though the world Pharmaceutical regulations are in continuous process of harmonization, they can be divided into four major categories based on the region, development strategy, regulations and marketing interest.
- North America (US, Canada)
- Europe (Europe Union, Eastern Europe)
- Rest of the World (Asia Pacific minus Japan, ANZ, GCC, LATAM, CEE, CIS,MCC)
- Japan
LATAM-Latin America; CEE-Central East Europe; CIS-Common Wealth of Independent States; ANZ-Australia, New Zealand; ROW-Rest of World; GCC-Gulf Cooperation Council.
Based on the economy and regulatory control of the countries, these are grouped into Regulated markets or Emerging markets. They not only differ by their region, but also in various other aspects like: how they regulate the pharmaceuticals, the different guidelines for registering the drugs, requirements to maintain the registrations, registration fee and patent regulations.
Table 1: List of Emerging Markets
|
S.NO. |
REGION |
AUTHORITY |
|
1. |
BRAZIL |
National Health Surveillance Agency (ANVISA) |
|
2. |
CHINA |
Chinese Food and Drug Administration (CFDA) |
|
3. |
RUSSIA |
Association of International Pharmaceutical Manufacturers. |
|
4. |
INDIA |
Central Drugs Standard Control Organization (CDSCO) |
Introduction to Generic Drug
A generic drug is a pharmaceutical product, usually intended to be interchangeable with a new drug (an innovator product) that is marketed after the expiry date of the patent or other exclusivity rights. A generic drug is defined as a “drug product that is similar to quantitative and qualitative composition of brand / reference listed drug product in dosage form, strength, route of administration, performance characteristics, and intended use.” Generic drugs are marketed under a non-proprietary or approved name rather than a proprietary or brand name.
Generic drugs are usually cheaper than the innovator drugs because of the following reasons:
1. No cost of identification and isolation of New Chemical Entity (NCE),
2. No cost of research and development,
3. Minimum marketing cost because branded drug is already approved as safe and effective.[2]
Role of Regulatory and Stages involved in Development of Generic Drugs
CHINA - Growing and Distinctive Pharmaceutical market
China is one of the largest pharmaceutical markets in the world, but the status is arguably due to the size of its population, as the market is not yet mature. The combined forces of economic and demographic development, government stimulus, and enhanced health awareness among the public, market consolidation, and improving R&D capability may help the country to grow into a more sophisticated market within the next decade.[3]
Chinese Regulatory Authority
The Regulatory Authority of People Republic of China is State Food and Drug Administration(CFDA)former it is termed as (SFDA). In March 2013, the regulatory body was re branded and restructured as the China Food and Drug Administration, elevating it to a ministerial-level agency. The CFDA replaced a large group of overlapping regulators with an entity similar to the Food and Drug Administrationof the United States, streamlining regulation processes for food and drug safety.The China Food and Drug Administration is directly under theState Council of the People's Republic of China, which is in charge of comprehensive supervision on the safety management of food, health food and cosmetics and is the competent authority of drug regulation in mainland China.
Steps involved in Registration of Imported Drugs in China
Generic product registration can be carried out by an agent in China. Overseas manufacturers of pharmaceutical products without legal representation in China are thus required to apply for product registration through agent services. An application is submitted to the CFDA, there by the review process is done by CDE (Center for Drug Evaluation).[4, 5]
General requirements for application dossiers
1. The first page of the dossiers shall be a directory for application items, which shall be arranged in order as per the “Provisions for Drug Registration” (SFDA Order No. 28). Each dossier shall indicate on its cover: the name of the drug and application item, document item number, and the name, phone number and address of contact person and the applicant.
2. All documents in the dossier shall be printed or copied in A4 size paper. The content shall be complete, standardized, clear, without alteration, and the data must be real and reliable.
3. The documents shall be put in portfolio envelope(s), on which the application classification, registration category, drug name, the envelope number of set X, the total number of envelopes in the set, original or copy, the contact person, phone number and the name of registration application agent shall be indicated.
4. Two sets of complete application dossiers (at least one set is in the original) and one set of review documents in hard copy shall be submitted for registration application. Four application forms (1 in the original and 3 in hard copy) shall be separately put into each set of dossier (the original application form and a hard copy shall be put in the set of the dossier in the original).
5. "Import Drug Registration Application Form": the drug registration application form submission program could be downloaded from CFDA website (cfda.gov.cn); the application form shall be filled in as required, printed and saved, and shall be signed by the overseas applicant, and signed & sealed by its domestic agent.
6. When mailing or submitting the application dossiers, the electronic version of the application form shall be sent to the following e-mail address dedicated for drug registration: slzx@cfda.gov.cn.
7. The data checking code on the electronic and paper application form should be identical.
8. Foreign language materials shall be translated into Chinese.[6]
Clinical Trial Application
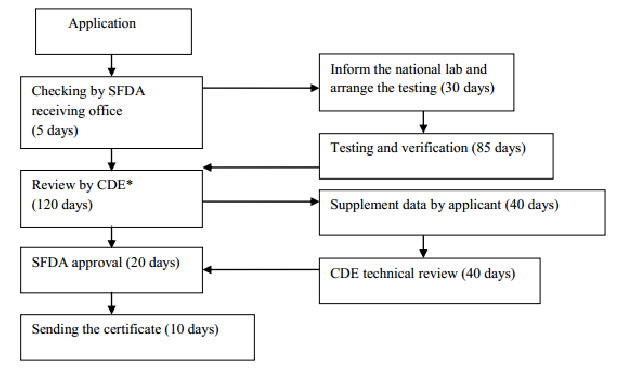
Flow chart 1: Imported drug registration process (Before clinical study)
Clinical Study
The Centre for Drug Evaluation will carry out a technical review of the test report and overall documentation which usually takes from 40 to 160 days to complete, depending on the product. The review report will be sent to the CFDA with recommendation on whether the product is subject to a clinical trial or bioequivalence study in China. Further information can be found in the Measures of the Administration of Drug Registration which outlines the requirements for clinical studies. If the Centre for Drug Evaluation deems that no clinical study is needed, the application will enter the final registration phase. In summary, the clinical study can be divided into 4 phases; however it is beyond this document to give a comprehensive introduction to clinical trials in China. Generally, phase I of the study needs between 20 to 30 subjects, phase II is approximately 200 subjects, phase III is 300 subjects, and phase IV is conducted as a post-marketing study investigating around 2000 subjects. For class III pharmaceutical products, a study with 100 pairs of subjects is required. For a bioequivalence study, generally 18-24 subjects are needed. Once the applicant receives approval for the clinical study, the applicant is free to choose the hospitals where the clinical study will be conducted from a list of designated clinical research hospitals or medical institutions listed on the CFDA website. It is a requirement that the clinical study needs to be conducted at a minimum of two different hospitals. The clinical study should be conducted in compliance with Good Clinical Practice (GCP).
All the pharmaceutical products used for the clinical study need to be tested, either by self-testing by the manufacturer or contracted to a designated testing laboratory coordinated by the NIFDC. After completing the clinical study, the clinical study plan, trial protocol, the approval documents by the Ethical Committee, together with patient consent forms and study report will form part of the drug registration application. It is difficult to give general statements about the timeframe for a Chinese clinical trial, as this will depend on availability of subjects, nature of disease, schedule of the hospital, etc. After completing the clinical study and pharmaceutical registration test, the applicant will need to fill in the drug registration form again and submit all documentation to the SFDA. The SFDAs Drug Evaluation Centre will review and evaluate all the submitted information. In some cases, the Drug Evaluation Centre will involve external experts in the evaluation of the pharmaceutical product. Once the Drug Evaluation Centre has passed its final judgment, the file is transferred to the SFDA for final approval. It is ultimately the decision of the SFDA to make its administrative decision for granting certification of the product or not. If the application is not approved, the applicant can apply for re-evaluation within 60 days. The pharmaceutical registration certificate is valid for 5 years and re-registration should be applied for at least 6 months prior to the certificate expiring. Re-registration should be submitted with all information of post-approval assessments in terms of the safety, efficiency and quality of the product done or collected within the 5 year validity period. In addition to the testing fee for the pharmaceutical registration test, and expenses for clinical trials, the drug registration fee amounts will be paid directly to the SFDA.[7-10]
Imported drug License
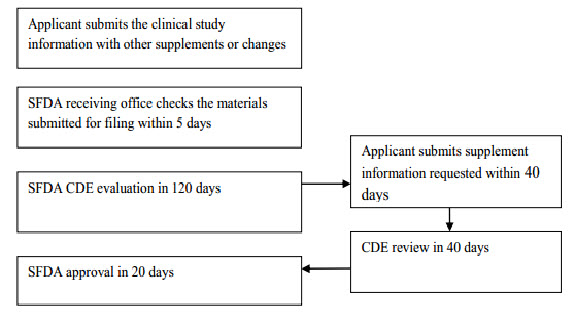
Flow chart 2: Imported drug registration process (after clinical study)
The first step in this process is to submit an application to the CFDA for a Clinical Trial Application (CTA). The CFDA will conduct a preliminary review of the submission package and then transfer the dossier to the Center for Drug Evaluation (CDE). Reviewers with background in pharmaceuticals, pharmacology and clinical study will run a technical review, while local sample testing will also be conducted in parallel. Few CTAs would pass through the CDE review in one single round. However, most applications will receive supplement notice(s) in writing to request additional information for further assessment. In such case, CDE will allow a 4-month period for applicant to gather and submit additional requested information to CDE. This entire CTA step usually takes at least 125 working days.
The second step in drug registration is Production Application (or Imported Drug License Application), which involves submitting a clinical report and other relevant dossiers to obtain an imported drug license. The process itself is basically the same as the CTA step. This second step will take approximately 145 days.[11]
Requirements for Approval of Imported Drugs in China[12]
Table 2: Requirements for Generic Imported Drug Submission in China
|
Dossier Format |
Regional specific, Recently initiated CTD format (still in initial phase) |
|
|
Data Exclusivity for Innovator products |
6 years |
|
|
Efficacy |
Clinical Trial is Mandatory |
|
|
Reference product |
Chinese Reference product |
|
|
Impact of patents on Submission Time Line |
Submitted before 2 years of patent expiry |
|
|
Certificate of pharmaceutical product |
Required for Submission |
|
|
DMF |
Yes |
|
|
Stability Batches |
3 BATCHES |
|
|
Stability Condition |
AT |
40°C ± 2°C ;75%RH ± 5%RH |
|
LT |
25°C ± 2°C ;60%RH ± 10%RH |
|
|
Stability Data Required at the Time of Submission |
AT = 6months LT = minimum 12 months |
|
|
Photo stability study |
Required on one batch |
|
|
Finished product Batch Testing Before Approval |
3 batches samples with quantity equivalent to 3 times the analysis |
|
|
Registration Validity Term |
Valid for 5 years |
|
Comparison of distinguishing parameters and requirements for Generic Drug approval process in China (Checklist)
Table 3: Requirements and Registration process of Generic drugs in China
|
PARAMETERS |
CHINA |
|
|
Regulatory Authority |
State Food and Drug Administration (CFDA) |
|
|
Web Address |
eng.sfda.gov.cn/ |
|
|
Climatic Zone |
Climatic ZoneII |
|
|
Language |
All information should be provided in Chinese and original language |
|
|
Dossier Format |
Regional specific, Recently initiated CTD format (still in initial phase) |
|
|
DMF Type |
DMF registration needed |
|
|
Reference Product |
Chinese Reference product |
|
|
Validity of License |
Valid for 5 years |
|
|
Clinical Trial Permission |
2-3 Years |
|
|
Import Drug Licence approval |
10-14 Months |
|
|
Application Forms |
NA |
|
|
Exhibit Batches |
3 Batches Samples |
|
|
Stability Studies Batches |
3 Batches |
|
|
Testing Frequency |
Accelerated |
6 months |
|
Long term |
minimum 12 months |
|
|
Stability Conditions |
Accelerated |
40°C ± 2°C ;75%RH ± 5%RH |
|
Long term |
25°C ± 2°C ;60%RH±10%RH |
|
|
Data Exclusivity |
6 Years |
|
|
Type of Submission |
Paper |
|
|
BA / BE Studies |
Clinical trials in China is Mandatory |
|
Summary
A generic drug is defined as a “drug product that is similar to quantitative and qualitative composition of brand / reference listed drug”. A Generic Product must meet the standards established by China Food and Drug Administration (CFDA) to be approved for marketing in China. This study covers the introduction to generic drugs in CHINA regulatory authority. It also includes the requirements and registration of Generic Drugs in above specified countries. & checklist for registration of generic drugs.
CONCLUSION
From the study we understoodthe regulations and requirements for generic drug registration in China.A Generic Product must meet the standards established by China Food and Drug Administration (CFDA) to be approved for marketing in China. Hence, we concluded the study by preparing a checklist which identifies the Registration requirements for generic drugs in China. Since China most emerging market, so there will be more scope for marketing of generic drugs in future. So one should focus on this market, but always consider the quality of API & always demand the DMF or technical file for API from API supplier/ Manufacturer from China while exporting our product to any country.
REFERENCES
1. Introduction to regulatory affairs. Published in Regulatory Affairs Professional Society (RAPS) [Internet]. RAPS; 2013 [cited 2014 Dec11]. Available from:
raps.org/uploadedFiles/Site_Setup/Home_Page/About_RAPS/The_Regulatory_Profession/PDF_Framework_Whitepaper.pdf
2. Introduction to Generic Drug. Published in WHO [Internet]. WHO; 2015 [cited 2014 Dec10]. Available from:
who.int/trade/glossary/story034/en/
3. CHINA - Growing and Distinctive Pharmaceutical market [Internet] CFDA [updated 2015 Feb; Cited on 2014 Dec 15]. Available from:
eng.sfda.gov.cn/WS03/CL0756/
4. Registration Categories of Drugs and Applications [Internet] CFDA; 2013 Dec 6 [Cited on 2014 Dec 18]. Available from: eng.sfda.gov.cn/WS03/CL0769/98158.html
5. Steps involved in Registration of Imported Drugs in China [Internet] CFDA; 2013 Dec 6 [Cited on 2014 Dec 17]. Available from:
eng.sfda.gov.cn/WS03/CL0769/98162.html
6. General requirements for application dossiers [Internet] CFDA; 2013 Dec 6 [Cited on 2014 Dec 12]. Available from:
eng.sfda.gov.cn/WS03/CL0766/61638.html#01
7. Specific Requirements for Chemical Drugs [Internet] CFDA; 2013 Dec 6 [Cited on 2014 Dec 10]. Available from:
eng.sfda.gov.cn/WS03/CL0769/98162.html
8. Standards and testing [Internet] CFDA; 2013 Dec 6 [Cited on 2014 Dec 11]. Available from: eng.sfda.gov.cn/WS03/CL0767/61640.html
9. Clinical Study [Internet] CFDA; 2013 Dec 6 [Cited on 2014 Dec 06]. Available from:
eng.sfda.gov.cn/WS03/CL0767/61639.html
10. Requirement for Local Registration of Clinical Trial[Internet] CFDA; 2013 Dec 6 [Cited on 2014 Dec 13]. Available from:
eng.sfda.gov.cn/WS03/CL0769/98158.html
11. Imported drug License [Internet] CFDA; 2013 Dec 6 [Cited on 2014 Dec 16]. Available from: eng.sfda.gov.cn/WS03/CL0768/98109.html
12. Requirements for Approval of Imported Drugs in China [Internet] CFDA; 2013 Dec 6 [Cited on 2014 Dec 18]. Available from:
eng.sfda.gov.cn/WS03/CL0769/98158.html
REFERENCE ID: PHARMATUTOR-ART-2387
|
PharmaTutor (Print-ISSN: 2394 - 6679; e-ISSN: 2347 - 7881) Volume 4, Issue 2 Received On: 07/09/2015; Accepted On: 11/09/2015; Published On: 01/02/2016How to cite this article: Badjatya JK; Regulatory requirements and registration process of Generic Drugs in China; PharmaTutor; 2016; 4(2); 13-18 |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






