

{ DOWNLOAD AS PDF }
ABOUT AUTHORS
Arijit Gandhi*1, Chandrani Roy2
1 Production cum Quality Manager, Kras Pharmaceuticals Pvt. Ltd., Fatwah, India.
2 Department of pharmaceutics, Gupta College of Technological Sciences, West Bengal
arijit.babugandhi.gandhi@gmail.com
ABSTRACT
Recently the concept of “Quality by Design” (QbD) gaining much attention among pharmaceutical industries for maintaining Quality. It serves as a bridge between industry and drug regulatory authorities to move towards a scientific, risk based, holistic and proactive approach for development of pharmaceutical product. It mainly covers designing and developing formulations and manufacturing processes to ensure predefined product quality. Some of the QbD elements include defining target product quality profile, designing product and manufacturing processes, identifying critical quality attributes, process parameters, and sources of variability & controlling manufacturing processes to produce consistent quality over time The purpose of this article is to discuss the concept of pharmaceutical Quality by Design and describe how it can be help to ensure pharmaceutical quality & drug development.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-2445
|
PharmaTutor (ISSN: 2347 - 7881) Volume 4, Issue 11 Received On: 15/06/2016; Accepted On: 29/06/2016; Published On: 01/11/2016 How to cite this article: Gandhi A, Roy C; Quality by Design (QbD) in Pharmaceutical Industry: Tools, Perspectives and Challenges; PharmaTutor; 2016; 4(11); 12-20 |
INTRODUCTION
Pharmaceutical industry is constantly searching the ways to ensure and enhance product safety, quality and efficacy. However, drug recalls, manufacturing failure cost, scale up issues and regulatory burden in recent past produce huge challenge for industry. In traditional, the product quality and performance are predominantly ensured by end product testing, with limited understanding of the process and critical process parameters. Regulatory bodies are therefore focusing on implementing quality by design (QbD), a science based approach that improves process understanding by reducing process variation and the enabling process-control strategies.
Instead of relying on finished product testing alone, QbD provides insights upstream throughout the development process. As a result, a quality issue can be efficiently analyzed and its root cause quickly identified. QbD requires identification of all critical formulation attributes and process parameters as well as determining the extent to which any variation can impact the quality of the finished product [1-3].
In the area of pharmaceutical quality; Food and drug administration (FDA) announced proposed amendments to "Current Good Manufacturing Practices" (cGMP) in 2002, with an emphasis on establishing a 21st century outlook on pharmaceutical manufacturing in order to establish a more systematic science and risk based approach to the development of pharmaceutical products. The initiation of the cGMPs for the 21st Century and the publication of the Process Analytical Technology (PAT) guidance in 2004 by the FDA gave the way for the modernization of the pharmaceutical industry. After that, ICH (International Conference on Harmonization) discussions in July 2003 (Brussels) agreed a consensus vision to develop a harmonized pharmaceutical quality system applicable across the life cycle of the product emphasizing an integrated approach to risk management and science. All the major objectives with regard to quality issues are being addressed by the ICH guidelines. The three ICH guidelines which throw light upon quality-by-design and related aspects include Q8 Pharmaceutical development, Q9 Pharmaceutical risk management and Q10 Pharmaceutical Quality systems. In fact, the ICH guideline Q8 is sub-divided into two parts: part one deals with pharmaceutical development and Part II is the annex to the guideline which states the principles for Quality-by-Design. According to ICH Q8(R2) guideline, Quality by Design (QbD) is “A systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and Process control, based on sound science and Quality Risk Management”[4-6].
QbD describes a pharmaceutical development approach referring to formulation design and development and manufacturing processes to maintain the prescribed product quality. Guidelines and mathematical models are used to ensure the establishment and use of the knowledge on the subject in an independent and integrated way [7]. In order to initiate a successful QbD program, the first step is to identify those process parameters that are essential to product quality and develop well – validated analytical methodologies to monitor those parameters. The objective of this review article is therefore to provide a comprehensive understanding on various aspects of QbD, along with addressing the concerns related to its implementation.
Key characteristics of QbD [8-9]
- A tool for focused & efficient drug development
- Dynamic and systematic process
- Relies on the concept that Quality can be built in as a continuum
- It is applicable to Drug Product and Drug Substance development (chemicals / biologics)
- It is applicable to analytical methods
- Can implemented partially or totally
- Can be used at any time in the life cycle of the Drug
- Always encouraged by Regulators.
Benefits of QbD [10-12]
- Eliminate batch failures
- Minimize deviations and costly investigations
- Avoid regulatory compliance problems
- Empowerment of technical staff
- Efficient, agile, flexible system
- Increase manufacturing efficiency, reduce costs and project rejections and waste
- Build scientific knowledge base for all products
- Better interact with industry on science issues
- Ensure consistent information
- Incorporate risk management
- Reduce end-product testing
- Speed-up release decision
[adsense:468x15:2204050025]
Key elements of QbD
ICH Q8: Pharmaceutical Development discusses the various elements of quality by design. These in combination with the enablers form the fundamental basis for the QbD approach to development. Figure 1 provides a pictorial representation of the typical elements of QbD.
It involves the following key elements during pharmaceutical development
- Define the Quality Target Product Profile
- Identify the Quality Attributes
- Perform a Risk (Assessment) Analysis
- Determine the Critical Quality Attributes and Critical Process Parameters
- Determine the Design Space
- Identify a Control Strategy

Fig 1. Overview of QbD.
Quality Target Product Profile (QTTP)
According to ICH Q8(R2), QTTP is “Prospective summary of the quality characteristics of a drug product that ideally will be achieved to ensure the desired quality, taking into account safety and efficacy of the drug product”. Basically it is a tool for setting the strategy for drug development. Recently QTTP is widely used in development planning, clinical and commercial decision making, regulatory agency interactions, and risk management.
It is the quality characteristics that the drug product should possess in order to reproducibly deliver the therapeutic benefit promised in the label. The QTTP guides formulation scientists to establish formulation strategies and keep the formulation effort focused and efficient. QTPP is related to identity, assay, dosage form, purity, stability in the label. For example, a typical QTPP of an immediate release solid oral dosage form would include
– Tablet Characteristics
– Identity
– Assay and Uniformity
– Purity/Impurity
– Stability, and
– Dissolution
It is important to acknowledge that QTPP should only include patient relevant product performance elements. For example, tablet density or hardness may be included as a specification for process monitoring but may not be included in QTPP. Also, if particle size is critical to the dissolution of a solid oral product, then the QTPP should include dissolution but not particle size [13-14].
Critical quality attributes (CQAs)
Once QTPP has been identified, the next step is to identify the relevant CQAs. A CQA is defined as “A physical, chemical, biological or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality”.
CQAs are generally associated with raw materials (drug substance, excipients), intermediates (in-process materials), and drug product. Drug product CQAs are the properties that are important for product performance, that is, the desired quality, safety, and efficacy (Fig.2).
This indicates that CQAs are subsets of QTPP that has a potential to be altered by the change in formulation or process variables [14-15]. For example, QTPP may include additional quality attributes of the drug product such as strength and dosage form, which are not the part of CQA as it will not change during drug development process. However, QTTP attributes such as assay, content uniformity, dissolution, and permeation flux will also be a part of CQA as they may be altered by formulation or process variables. For example, the CQAs of drug substance and drug product are enlisted in Table 1.
Identification of CQAs is done through risk assessment as per the ICH guidance Q9. Prior product knowledge, such as the accumulated laboratory, nonclinical and clinical experience with a specific product-quality attribute, is the key in making these risk assessments. Such knowledge may also include relevant data from similar molecules and data from literature references. Taken together, this information provides a rationale for relating the CQA to product safety and efficacy [16].

Fig. 2. Decision Tree to Decide CQAs.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Table 1. Typical CQAs for drug substance and drug products.
|
For Drug Substance (chemical) |
For Drug product (tablet) |
|
|
Quality risk management (QRM)
The FDA defines a Risk Management as, a strategic safety program designed to decrease product risk by using one or more interventions or tools. It is systematic process for the assessment, control, communication and review of risks to the quality of the drug product across the product lifecycle [17]. Overview of a typical quality risk management process is given in Fig. 3.
The ICH Q9 guideline: Quality Risk Management provides a structure to initiate and follow a risk management process. The relevant tools of QRM are as follows:
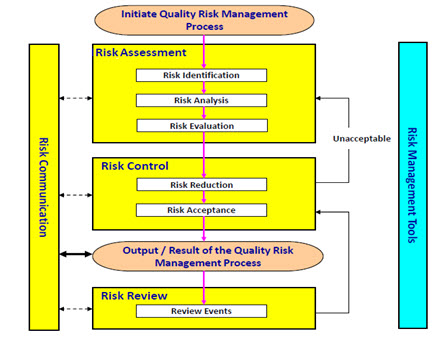
Fig. 3. Overview of a typical quality risk management process (as per ICH Q9: Quality Risk Management).
Failure mode effects analysis (FMEA)
FMEA is one of the most commonly used risk-assessment tools in the pharmaceutical industry. It is a systematic and proactive method to identify and mitigate the possible failure in the process. Failure modes represent any errors or defects in a process, material, design, or equipment. Once failure modes are established, FMEA tool evaluates the effect of these failures and prioritizes them accordingly. This tool is further advanced with studying criticality of the consequences and providing clear indication of situation.
Failure Mode, Effects and Criticality Analysis (FMECA)
It is the extension of earlier said FMEA tool. Extending FEMA to incorporate an investigation of the degree of severity of consequences, their probabilities of occurrence and their detect-ability is Failure mode, effects and criticality analysis. In FMECA, each failure mode of the product is identified and then evaluated for criticality. This criticality is then translated into a risk, and if this level of risk is not acceptable, corrective action must be taken. This can be utilized for failure and risk associated with manufacturing processes. The tool can also be used to establish and optimize maintenance plans for repairable systems and/or contribute to control plans and other quality assurance procedures.
Fault tree analysis (FTA)
This tool assumes failure of the functionality of a product or process. The results are represented pictorially in the form of a tree of fault modes. This can be used to investigate complaints or deviation in order to fully understand their root cause and ensure that intended improvement will resolve the issues and not cause any other different problem.
Hazard analysis and critical control points (HACCP)
HACCP provides detailed documentation to show process or product understanding through identifying parameters to control and monitor. The definition of hazard includes both safety and quality concern in a process or product. It involves hazard analysis, determining critical control point, establishing critical limit, establishing a system to monitor critical control point and establishing a record keeping system. This might be used to identify and manage risk associated with physical, chemical and biological hazards.
The output of a risk assessment may be a combination of quantitative and qualitative estimation of risk. As part of FMEA, a risk score or Risk Priority Number (RPN) may be assigned to the deviation or to the stage of the process that is affected; this helps to categorize the deviation. RPN is calculated by multiplying Probability (P), Detectability (D) and Severity (S), which are individually categorized and scored. Rating scales usually range from 1 to 5.
RPN = probability score × severity score × detectability score
Where, the score was defined prior to the risk analysis stage. A RPN of < 40 was considered a low risk; a RPN of 40–99 was identified as an intermediate risk; and a RPN of ≥ 100 was defined as a high risk [14, 17].
Determination of Critical Process Parameters
A critical process parameter (CPP) is any measurable input (input material attribute or operating
parameter) or output (process state variable or output material attribute) of a process step that must be controlled to achieve the desired product quality and process consistency. A parameter is critical when a realistic change in that parameter can cause the product to fail to meet the QTPP. Thus, whether a parameter is critical or not depends on how large of a change one is willing to consider. Thus the first step in classifying parameters is to define the range of interest which we call the potential operating space (POS). The POS is the region between the maximum and minimum value of interest for each process parameter. Criteria for identifying critical and non-critical parameters are that a parameter is non-critical when there is no trend to failure within the POS and there is no evidence of interactions within the proven acceptable range (PAR), which is the range of experimental observations that lead to acceptable quality [18]. The different CCPs during tablet manufacturing along with CQAs are given in Table 2.
Table 2. Different critical process parameters with potential quality attributes during tableting.
|
Operations during tableting |
Critical Process Parameters |
Potential quality attributes |
|
Wet granulation |
|
|
|
Drying |
|
|
|
Milling |
|
|
|
Mixing |
|
|
|
Compression |
|
|
|
Coating |
|
|
Design space
ICH Q8(R2) defines design space as “ the multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters that have been demonstrated to provide assurance of quality. Working within the design space is not considered as a change. Movement out of the design space is considered to be a change and would normally initiate a regulatory post approval change process. Design space is proposed by the applicant and is subject to regulatory assessment and approval.”
Design space may be constructed for a single unit operation, multiple unit operations, or for the entire process. Though according to FDA guideline, defining design space is optional since the product and process understanding can be established without a formal design space, nevertheless, such approach can assist to better understanding and attain overall control of a system.
The Design Space is linked to criticality through the results of risk assessment, which determines the associated CQAs and CPPs. It describes the multivariate functional relationships between CQAs and the CPPs that impact them, and should include their linkage to or across unit operations. Such relationships are arrived at by iterative application of risk assessment and experimental design, modeling, as well as the use of literature and prior experience.
Methods for determining design space included: one-variable-at-a-time experiments, statistically designed experiments, and modeling approaches. Methods for presenting design space included graphs (surface-response curves and contour plots), linear combination of parameter ranges, equations, and models. Alternatively, the design space can be explained mathematically through equations describing relationships between parameters for successful operation [19-21].
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Control Strategy
ICH Q10 defines a control strategy as “a planned set of controls derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to drug substance and drug product materials and components, facility and equipment operating conditions, in process controls, finished product specifications and the associated methods and frequency of monitoring and control.” A control strategy normally include input material controls, process controls and monitoring, design space around individual or multiple unit operations, and/or final product specifications used to ensure consistent quality [22, 23]. The finished drug products are tested for quality by assessing if they meet specifications. In addition, manufacturers are usually expected to conduct extensive in process tests, such as blend uniformity or tablet hardness.
A QbD based control strategy for blending process is shown in Fig. 4. Pharmaceutical quality is assured by understanding and controlling formulation and manufacturing variables to assure the quality of the finished product. The end product testing only confirms the quality of the product.
Fig. 4. Example of control strategy for QbD process.
Challenges
Though Quality by design is an essential part of the modern approach to pharmaceutical quality, but Lack of understanding regarding the pharmaceutical process is the cause and also the major limitation for QbD implementation. Pharmaceutical companies are traditionally tuned to care more about the end product, with little emphasis on the science-based understanding of the process involved. The majority of pharmaceutical companies feel that there is a need for a more easy guidance on how to actually implement QbD. Companies wanted clarification from FDA on QbD terminologies, acceptable methods, criteria to select and deselect critical quality attributes, standards by which to judge adequacy of controls, and criteria for analytical method substitution [23, 24]. 10 key challenges are the most problematic for QbD adoption. These challenges are evaluated by their relevancy against different drug types as well as different levels of adoption.
The first four challenges occur within companies:
- Internal misalignment (Disconnect between cross functional areas, e.g., R&D and manufacturing or quality and regulatory)
- Lack of belief in business case i.e. there is a lot of uncertainty over timing of and investment requirements for QbD implementation.
- Lack of technology to execute (e.g., Difficulty managing data, limited understanding of Critical Quality Attribute (CQA) implications)
- Alignment with third parties (i.e., How to implement QbD with increasing reliance on suppliers and contract manufacturers?)
The next six challenges are directly related to the regulatory authority:
- Inconsistency of treatment of QbD across regulatory authority
- Lack of tangible guidance for industry
- Regulators not prepared to handle QbD applications
- The way promised regulatory benefits are currently being shared does not inspire confidence
- Misalignment of international regulatory bodies
- Current interaction with companies is not conducive to QbD
It is accepted that the challenges and concerns associated with the implementation of QbD can only be resolved if there is efficient communication between the industry and the regulatory bodies.
CONCLUSIONS
QbD is increasingly becoming an important and widely used technique in pharmaceutical product development. While QbD is most effective when it is employed at a product/process design level, it should also be accomplished in the manufacturing and quality assurance environments. Implementing QbD concept in product development provide quality medicines to patients, production improvements to Manufacturers with significantly reduced batch failures and drug regulatory bodies will have greater confidence in the robust quality of products. This approach allows the establishment of priorities and flexible boundaries in the process. As such QbD is becoming a promising scientific tool in quality assurance in pharmaceutical industry.
REFERENCES
1. Woodcock J, The concept of pharmaceutical quality. American Pharmaceutical Review 2004;7: 10–15.
2. Lionberger RA, Lee LS, Lee L, Raw A,Yu LX. Quality by design: Concepts for ANDAs. The AAPS Journal 2008; 10: 268–276.
3. Looby M, Ibarra N, Pierce JJ, Buckley K, O'Donovan E, Heenan M. Application of quality by design principles to the development and technology transfer of a major process improvement for the manufacture of a recombinant protein. Biotechnology Progress 2011;27:1718-29.
4. Q9: Quality Risk Management. ICH Harmonized Tripartite Guidelines. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2006.
5. Q10: Pharmaceutical Quality System, ICH Tripartite Guidelines. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2007.
6. Q8 (R1): Pharmaceutical Development, Revision 1, ICH Harmonized Tripartite Guidelines, International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2007.
7. Yu LX, Pharmaceutical quality by design: Product and process development, understanding, and control. Pharmaceutical Research 2008; 25: 781–791.
8. Gupta A, Fuloria NK. Short review on Quality by design: A new Era of Pharmaceutical drug development. Int. J. Drug Dev. & Res. 2012; 4:19-26.
9. Elliott P, Billingham S, Bi J, Zhang H. Quality by design for biopharmaceuticals: a historical review and guide for implementation. Pharmaceutical bioprocessing 2013; 1:105-122.
10. Nadpara NP, Thumar RV, Kalola VN, Patel PB. Quality By Design (QbD) : A Complete Review. Int. J. Pharm. Sci. Rev. Res. 2012; 17: 20-28.
11. Pohl M, Schweitzer M, Hansen G, Hanna BM, Borman P, Smith K, Larew J, Nethercote P. Implications and opportunities of applying the principles of QbD to analytical measurements. Pharm. Technol. Eur. 2010; 22: 29–36.
12. Eziokwu NV. Quality by design (QbD): manufacturing and product quality of Generics drugs perspective. Journal of Global Trends in Pharmaceutical Sciences 2013;4: 1257-1262.
13. Yu LX, Lionberger R, Olson MC, Johnston G, Buehler G, Winkle H. Quality by Design for Generic Drugs. Pharmaceutical Technology 2009;33:122-27.
14. Patil AS, Pethe AM. Quality by Design (QbD) : A new concept for development of quality pharmaceuticals. International Journal of Pharmaceutical Quality Assurance 2013; 4: 13-19.
15. Nagar M, Panwar KS, Chopra VS, Bala I, Triv P. Quality by design: A systematic approach to pharmaceutical development. Der Pharmacia Lettre 2010; 2: 111-130.
16. Kumar VP, Gupta NV. A Review on quality by design approach (QBD) for Pharmaceuticals. Int. J. Drug Dev. & Res. 2015; 7: 52-60.
17. Bhattacharya J. Quality Risk Management –Understanding and Control the Risk in Pharmaceutical Manufacturing Industry. International Journal of Pharmaceutical Science Invention 2015; 4: 29-41.
18. Chowdary KPR, Ravi Shankar K, Kumar PS. Recent research on QbD approach in formulation development - a review. International Journal of Chemical Science and Technology 2014; 4: 282-292.
19. Wu H, White M, Khan MA. Quality-by-Design (QbD): An integrated process analytical technology (PAT) approach for a dynamic pharmaceutical co-precipitation process characterization and process design space development. Int J Pharm. 2011;405:63-78.
20. Altan S, Bergum J, Pfahler L, Senderak E, Sethuraman S, Vukovinsky KE. Statistical Considerations in Design Space Development (Part I of III). Pharmaceutical Technology 2010;34: 66-70.
21. Altan S, Bergum J, Pfahler L, Senderak E, Sethuraman S, Vukovinsky KE. Statistical Considerations in Design Space Development (Part II of III). Pharmaceutical Technology 2010; 34: 52-60.
22. Trivedi B. Quality by desing (QbD) in pharmaceuticals. Int J Pharm Pharm Sci. 2012; 4:17-29.
23. Jain S. Quality by design (QbD): a comprehensive understanding of implementation and challenges in pharmaceuticals development. Int J Pharm Pharm Sci. 2013; 6: 29-35.
24. Drakulich, A. Critical challenges to implementing QbD: A Q&A with FDA. Pharm. Technol. 2009; 33: 90–94.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






