

{ DOWNLOAD AS PDF }
ABOUT AUTHORS:
Chaudhary Sonam*, Rathore KS
Department of Pharmaceutics,
Bhupal Noble’s Institute of Pharmaceutical Sciences,
Sewashram road, Udaipur, Rajasthan, India
*chaudharysonam1993@gmail.com
ABSTRACT
A new approach to drug development could increase efficiencies, provide regulatory relief, flexibility, and offer important business benefits throughout the product’s life cycle. Quality by design is a systemic approach and essential part of the modern approach for quality and pharmaceutical development.
It includes defining target product quality profile, designing product, developing formulations, manufacturing processes, identifying critical quality attributes, process parameters, sources of variability and controlling manufacturing processes to ensure consistent product quality over time.
Pharmaceutical quality can be assured by understanding, controlling formulation and manufacturing variables using Quality by design. Product quality can be confirmed by product testing. Pharmaceutical industry have to work hard to develop, manufacture, to bring new drugs to market, to comply with regulatory requirements to ensure that the drugs are safe and effective. Implementation of Quality by design leads to transformation of the chemistry, manufacturing, and controls (CMC) review of abbreviated new drug applications (ANDAs) into a science-based pharmaceutical quality assessment.
INTRODUCTION1-5
“Quality by design means designing and developing manufacturing processes during the product development stage to consistently ensure a predefined quality at the end of the manufacturing process.”
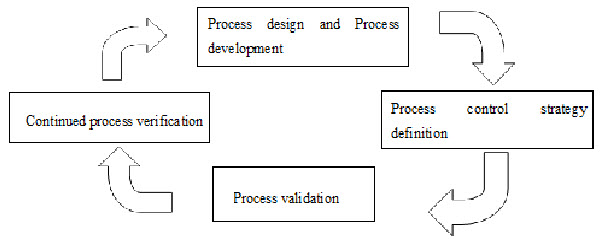
Fig.1: Quality by Design2
The concept of QbD was mentioned in the ICH Q8guideline, which states that “quality cannot be tested into products i.e., quality should be built in by design.” According to ICH Q8 QbD is defined as a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management.
Quality by Design (QbD) has become a new concept for development of quality pharmaceutical products, It is an essential part of the modern approach to pharmaceutical quality, QbD is a best solution to build a quality in all pharmaceutical products but it is also a major challenge to the Pharmaceutical industry whose processes are fixed in time, despite inherent process and material variability, Under this concept of QbD throughout designing and development of a product, it is essential to define desire product performance profile [Target product Profile (TPP), Target Product Quality Profile (TPQP)] and identify critical quality attributed (CQA). On the basis of this we can design the product formulation and process to meet the product attributes. This leads to recognize the impact of raw materials [critical material attributes (CMA), critical process parameters (CPP) on the CQAs and identification and control sources of variability. QbD is an emerging idea which offers pharmaceutical manufacturer with increased self-regulated flexibility while maintaining tight quality standards and real time release of the drug product.
ICH 2,3,6,7
ICH Q8, Q9, Q10 & Q11are designed as separate but linked in a series of documents exploring pharmaceutical products lifecycle
ICH Q8 Pharmaceutical Development
ICH Q9 Quality Risk Management
ICH Q10 Pharmaceutical Quality System
ICH Q11 - Development and Manufacture of Drug Substance.
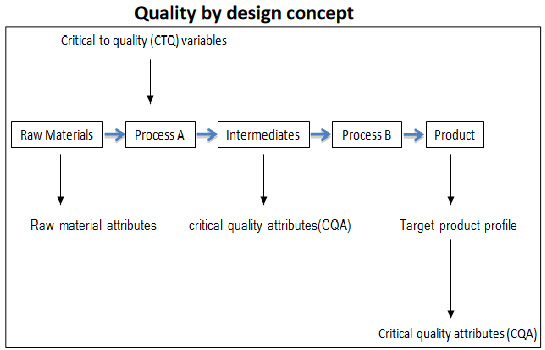
Fig.2: Concept of QbD3
Quality by design system
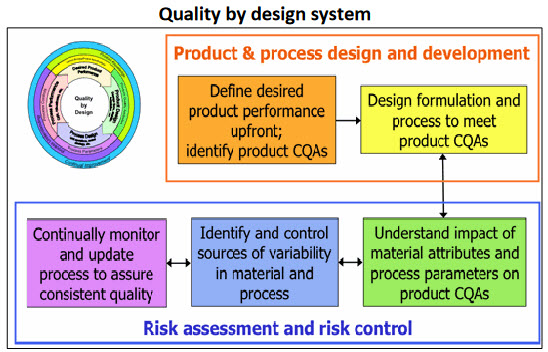
Fig.3: QbD system7
Steps 8,9,10
a. Development of new molecular entity
· Preclinical study.
· Nonclinical study.
· Clinical Study.
· Scale up.
· Submission for market Approval
b. Manufacturing
· Design Space.
· Process Analytical Technology.
· Real time Quality Control.
c. Control strategy
· Risk based decision.
· Continuous Improvement.
· Product performance
Benefits of QbD 1,3,5,6
1. QbD is good Business.
2. Eliminate batch failures.
3. Minimize deviations and costly investigations.
4. Avoid regulatory compliance problems.
5. Organizational learning is an investment in the future.
6. QbD is good Science.
7. Better development decisions.
8. Empowerment of technical staff.
Opportunities 8,9
1. Efficient, agile, flexible system.
2. Increase manufacturing efficiency; reduce costs and project rejections andv waste.
3. Build scientific knowledge base for all products.
4. Better interact with industry on science issues.
5. Ensure consistent information
6. Incorporate risk management.
Process [11, 12, 13]
Quality by Design is a scientific risk based holistic and proactive approach to pharmaceutical development. It involves the designing and planning of a drug product and process before actual experiment. Over the past several years, pharmaceutical scientists have provided several more specific definitions of the elements of quality by design and a draft of an annexure to ICH Q8 has been released.
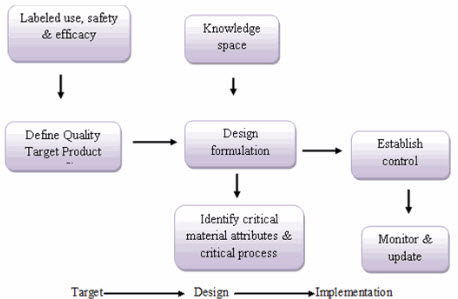
Fig.4: Process[11]
Elements
· Identifying Target Product Quality Profile (TPQP).
· Identifying CQA’s.
· Design product and defining product design space.
· Process design and defining process design space.
· Defining control strategy.
· Process validation.
· Regulatory filings.
· Process monitoring, life cycle management and continuous improvement.
Quality by Design Tools
Target product profile(TPP).14, 15
The role of target product profile (TPP) is to serve as a tool for “quality planning” for the drug product with “the end in mind” i.e. a summary of the drug development program described in the context of prescribing information goals. A quality target product profile (QTPP) is a term which is a natural extension of TPP for product quality .A QTPP relates to the quality of a drug substance or the drugs products that is necessary to deliver a desired therapeutic effect .QTPP is a predetermined summary of the characteristics of the drug product that will ideally be essential to ensure the desired quality with respect to safety and efficacy of the product. These predetermined QTPP evolve over time during drug development and may be modified to incorporate new knowledge, as is warranted by ongoing clinical studies such a dose effect and toxicology data.
Critical quality attributes (CQA).
Critical quality attributes are defined as physical, chemical, biological or microbiological properties or characteristics that need to be controlled to ensure product quality.( According to ICH Q8) CQAs as physical, chemical, biological or microbiological properties or characteristics that should be within an appropriate limit, range, or distribution to ensure the desired product quality. CQA has been used by some to describe elements of the TPQP while others have used CQA to describe mechanistic factors that determine product performance. Thus CQA is used to describe both aspects of product performance and determinants of product performance.
Risk assessment16, 17, 18
A key objective of risk assessment in pharmaceutical development is to identify which material attributes and process parameters affect the drug product CQAs, that is, to understand and predict sources of variability in the manufacturing process so that an appropriate control strategy can be implemented to ensure that the CQAs are within the desired requirements.
The identification of critical process parameters (CPP) and critical material attributes is an iterative process and occurs throughout development. During the initial phases of development, prior knowledge serves as the primary basis for the designation as there is not sufficient process/product understanding on the product under development. Therefore, the risks identified at the initial phases are perceived risks and as further process/product understanding is gained, the actual risks become clearer and a control strategy can be better defined. The risk assessment tools used in earlier phases of development therefore tend to be more qualitative and serve as a means to prioritize the experimentation.
Design space
ICH Q8 defines design space as, the multidimensional combination and interaction of input variables (material attributes) and process parameters that have been demonstrated to provide assurance of quality. Moving out of the design space is considered to be a change and would normally initiate a regulatory post-approval change process. The design space is proposed by the applicant and is subject to regulatory assessment and approval. Design space is potentially scale and equipment dependent, the design space determined on the laboratory scale may not be relevant to the process at the commercial scale. Therefore, design-space verification at the commercial scale becomes essential unless it is confirmed that the design space is scale-independent. Currently, generic drug sponsors obtain information about acceptable ranges for individual CPPs and CMAs at laboratory or pilot scales.
Multidimensional combination of and interaction of input variables and process parameters that have been demonstrated to provide Quality Assurance
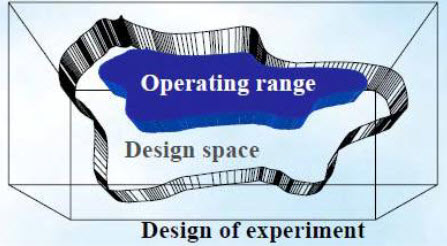
Fig.5: Design space17
Control Strategy19, 20
A planned set of controls, derived from current product and process understanding that ensures process performance and product quality…..” ICH Q8 (R2) & Q10
Control Strategy includes following elements (but not limited to):
· Input material attributes (e.g. drug substance, excipients, container closure).
· Equipment operating conditions (process parameters).
· In-process controls.
· Finished product specifications.
· Controls for each unit operations.
· Methods and frequency of monitoring and control.
Life cycle Management and Continuous improvement20-23
After approval, CQAs are monitored to ensure that the process is performing within the defined acceptable variability that served as the basis for the filed process design space. The primary benefit of an expanded process design space would be a more flexible approach by regulatory agencies. In the QbD paradigm, process changes within the design space will not require review or approval. Therefore, process improvements during the product life cycle with regard to process consistency and throughput could take place with fewer post approval submissions. In addition to regulatory flexibility, the enhanced understanding of the manufacturing process would allow more informed risk assessment as per ICH Q9 regarding the affects of process changes and manufacturing deviations on product quality. Manufacturing, the experience grows and opportunities for process improvement identified are operating space could be revised within the design space without the need for post-approval submission. Over the lifetime of a product, process changes may be required to be made and may require process characterization, validation and filing of the changes to the approved process design space. The quality system needs to provide adequate oversight during QbD implementation of changes that will not go through regulatory approval. Robustness of the quality system would need to be demonstrated with respect to the following four elements: process performance/product quality monitoring; preventative/corrective action, change management and management review of process performance and product quality
Problems in implementing QbD
1. Internal misalignment.
2. Lack of belief in business case.
3. Lack of technology to execute.
4. Alignment with third parties.
5. Inconsistency of treatment of QbD across FDA.
6. Lack of tangible guidance for industry.
7. Regulators not prepared to handle QbD application.
8. The way promised regulatory benefits are currently being shared dose not inspire confidence.
9. Misalignment of international regulatory bodies.
10. Current interaction with companies is not conducive to QbD.
CONCLUSION
The goal of QbD is to reduce product variability and defects, thereby enhancing patient efficacy and safety. Furthermore, it also helps in enhancing the product and process development depth and understanding which then directly improves the efficiency and helps to effectively manage the post approval changes. The elements QbD-QTPP, CQA, CMA and CPP enhance the product development process leading to a thorough product and process understanding, successful scale-up, control strategy and continual improvement. Finally, product and process capability is continually reviewed and improved post approval during product lifecycle management.
REFERENCES
1. Woodcock J, The concept of pharmaceutical quality. American Pharmaceutical Review, 7(6), 2004, p.10–15.
2. Q9: Quality Risk Management. ICH Harmonized Tripartite Guidelines International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2006.
3. Q10: Pharmaceutical Quality System, ICH Tripartite Guidelines. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2007.
4. Lionberger RA, Lee LS, Lee L, Raw A ,Yu LX, Quality by design: Concepts for ANDAs, The AAPS Journal, 10, 2008, p.268–276.
5. FDA Guidance for Industry and Review Staff: Target Product Profile – A Strategic Development Process Tool (Draft Guidance).
6. Q8 (R1): Pharmaceutical Development, Revision 1, ICH Harmonized Tripartite Guidelines, International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2007.
7. Food and Drug Administration, Office of Generic Drugs White Paper on Question-based Review: fda.gov/cder/ OGD/QbR.htm.
8. Callis JB, Illman DL, Kowalski BR, Process analytical chemistry. Analytical Chemistry, 59, 1987, 624A–637A.
9. Yu LX, Pharmaceutical quality by design: Product and process development, understanding, and control. Pharmaceutical Research, 25, 2008, 781–791.
10. Munson J, Gujral B, Stanfield CF, A review of process analytical technology (PAT) in the U.S. pharmaceutical industry. Current Pharmaceutical Analysis, 2, 2006, 405–414.
11. Juran JM. On quality by design. The new steps for planning quality into goods and services New York free press 1992 p.no.1-2.
12. MN Nasr. Implementation of quality by design (QbD): status, challenges, and next steps. FDA Advisory Committee for Pharmaceutical Science. Available at: fda.gov/ohrms/dockets/ac/06/slides/2006-4241s1_6.ppt (accessed 9/7/2015).
13. ISPE PQLI. Draft PQLI summary update report. . Org/cs/pqli_product_quality_lifeycle_implementation_/draft_pqli_summary_Update- report (accessed 11/21/2007).
14. Food and Drug Administration: fda.gov/ohrms/ dockets/ac/06/minutes/2006–4228m1.pdf, 2006.
15. US Food and Drug Administration, Guidance for Industry: Q10 Pharmaceutical Quality Systems, 2009.
16. Guidance for industry “Q9 Quality Risk Management” by US department of Health and Human Services, Food and drug Administration, Center for Drug and Evaluation Research, June 2006.
17. WHO Guideline on Quality Risk Management A draft guidance August 2010.
18. Implementation of ICH Q9 in the pharmaceutical field an example of methodology from PIC/S 2010.
19. Gibson, M, Product Optimization: Pharmaceutical Preformulation and Formulation. Taylor & Francis, New York 2001.
20. Seely, RJ, Haury, J, in: Rathore, AS, Sofer, G (Eds.), Process Validation in Manufacturing of Biopharmaceuticals, Taylor & Francis, Boca Raton, FL 2005, pp.13-50 .
21. US Food and Drug Administration. guidance for industry: Q10 quality systems approach to pharmaceutical cGMP regulations (FDA, Rockville, MD, September 2006)
22. US Food and Drug Administration. Pharmaceutical cGMPs for the 21st century: a risk-based approach (FDA, Rockville, MD, August 2002). fda. gov/oc/guidance/gmp.html.
23. Rathore KS, Pokhrana D, Yadav S, Quality by Design, articlesbase.com/college-and-university-articles/quality-by-design-qbd-4890152.html (accessed on July4, 2015).
REFERENCE ID: PHARMATUTOR-ART-2376
|
PharmaTutor (Print-ISSN: 2394 - 6679; e-ISSN: 2347 - 7881) Volume 3, Issue 12 Received On: 06/06/2015; Accepted On: 16/08/2015; Published On: 01/12/2015How to cite this article: S Chaudhary, KS Rathore; Quality by Design; PharmaTutor; 2015; 3(12); 23-28 |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






