

{ DOWNLOAD AS PDF }
ABOUT AUTHORS:
Pankaj Dinkar Patil*, Raju Tayade
Department of Pharmaceutical sciences and technology,
ICT Mumbai, Maharashtra
pankaj.patil46@gmail.com
ABSTRACT:
Tumor hypoxia is feature of locally advancing solid tumor and also considered as potential therapeutic challenge. It causes solid tumor resistible to the cancer treatment like radiation and chemotherapeutic drugs. The transcription factor HIF-1 is the major player in the regulation of tumor progression into malignant phenotype. We can target this hypoxic conditions using pathophysiological approach for targeted drug delivery for treatment of cancer. This review will focus on various biological aspects of tumor hypoxia necrosis and approaches for targeting hypoxic tumors. This tumor selective treatment will include bioreductive drugs, gene therapy, and recombinant bacteria.
REFERENCE ID: PHARMATUTOR-ART-2201
INTRODUCTION:
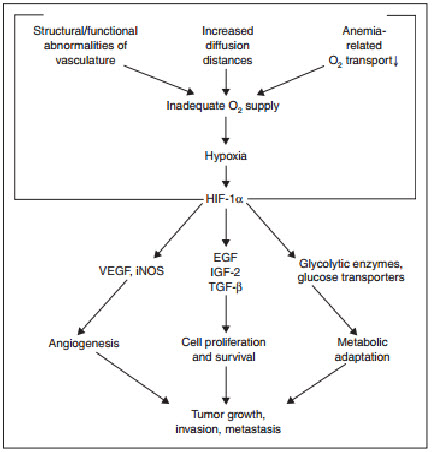
Hypoxia (also called as Hypoxiation or Anoxemia) is a condition in which the body or a region of the body is deprived of adequate oxygen supply. Hypoxia may be classified as either systemic, affecting the whole body, or local, affecting a region of the body. Hypoxia differs from hypoxemia in that hypoxia refers to a state in which oxygen supply is insufficient, whereas hypoxemia refers specifically to states that have low arterial oxygen supply. Hypoxia is a characteristic feature of locally advanced solid tumors resulting from an imbalance between oxygen (O2) supply and consumption[1]. The presence of hypoxic regions of low levels of oxygen in human tumors was postulated by Thomlinson and Gray some 50 years ago based on their observations of the distribution of necrosis relative to blood vessels.It was known at that time that hypoxic cells were resistant to killing by ionizing radiation, and this led to clinical trials with patients undergoing radiotherapy in hyperbaric oxygen chambers, to try to force more oxygen into the blood and into the tumor. Solid tumors comprise approximately 90% of all known cancers like Breast cancer, Prostate cancer, Lung cancer etc. They develop from a single mutated cell and leadto significant morbidity and mortality, either by invading normal tissue or by metastasizing to vital organs, such as the liver, lung, or brain. The process of tumor progression (i.e., proliferation, local invasion, and distant metastasis) is characterized by rapid cellular growth accompanied by alterations of the microenvironment of the tumor cells.In solid tumors, oxygen delivery to the respiring neoplastic and stromal cells is frequently reduced or even abolished by deteriorating diffusion geometry, severe structural abnormalities of tumor micro vessels, and damaged microcirculation. Sustained hypoxia in a growing tumor may cause cellular changes that can result in a more clinically aggressive phenotype. During the process of hypoxia-driven malignant progression, tumors may develop an increased potential for local invasive growth, perifocal tumor cell spreading, and regional and distant tumor cell spreading.
To grow beyond a diameter of approximately 1 mm, newly developing tumors must form their own vascular network and blood supply, which they accomplish either by incorporating preexisting host vessels or by forming new microvessels through the influence of tumor angiogenesis factors. However, the newly formed vascular network differs greatly from that found in normal tissue, typically displaying a broad range of structural and functional abnormalities, including dilations, incomplete or absent endothelial linings and basement membranes, leakiness, irregular and tortuous architecture, arteriovenous shunts, blind ends and a lack of contractile wall components and pharmacological/physiological receptors. These abnormalities lead to irregular and sluggish blood flow, therebydiminishing the delivery of O2(and nutrients) to the tumor cells, with the resultant development of hypoxic or even anoxic areas. The oxygenation status of the tumor can be worsened further by increases in diffusion distances, which occur when the tumor cells spread beyond the distance that allows adequate delivery of O2 by the blood vessels (>70 µm)[2].
For many years, tumor hypoxia has been recognized as a potential therapeutic problem because of its adverse impact on the effectiveness of radiation therapy. However, hypoxia has recently emerged as a major factor that influences tumor proliferation and malignant progression. Additionally, hypoxia may induce downregulation of adhesion molecules, thereby facilitating tumorcell detachment. Hypoxia-induced or hypoxia-mediated changes of the proteome (i.e., the complete set of proteins within a cell at a given time) of the neoplastic and stroma cells and the genome of the genetically unstable neoplastic cells may explain the fact that tumor oxygenation is associated with disease progression, a link that has been demonstrated for a variety of human malignant tumor types. The aim of this review is to compile current details of hypoxia and the phenomena of malignant progression and resistance toward oncologic treatment and how one can exploit pathophysiological conditions for targeted drug delivery [3].
DEFINITION AND CAUSES OF HYPOXIA:
Tissue hypoxia results from the inadequate supply of oxygen that compromises biologic functions. Oxygen passively diffuses in the lung alveoli according to a pressure gradient. Oxygen diffuses from the breathed air, mixed with water vapour, to arterial blood, where its partial pressure is around 100 mmHg (13.3kPa). In the blood, oxygen is bound to hemoglobin, a protein in red blood cells. The binding capacity of hemoglobin is influenced by the partial pressure of oxygen in the environment, as described in the oxygen–hemoglobin dissociation curve. A smaller amount of oxygen is transported in solution in the blood.In peripheral tissues, oxygen again diffuses down a pressure gradient into cells and their mitochondria, where it is used to produce energy in conjunction with the breakdown of glucose, fats and some amino acids.Hypoxia can result from a failure at any stage in the delivery of oxygen to cells[3]. This can include decreased partial pressures of oxygen, problems with diffusion of oxygen in the lungs, insufficient available hemoglobin, problems with blood flow to the end tissue, and problems with breathing rhythm. Experimentally, oxygen diffusion becomes rate limiting (and lethal) when arterial oxygen partial pressure falls to 40 mmHg (5.3kPa) or below.
Causes of hypoxia:
Ischemia
Ischemia meaning insufficient blood flow to a tissue can also result in hypoxia. This is called 'Ischemic hypoxia’. An example of insufficient blood flow causing local hypoxia is gangrene that occurs in diabetes.
Hypoxemic hypoxia
It is specific hypoxic state where the arterial content of oxygen is insufficient.
Problems with hemoglobin
Almost all the oxygen in the blood is bound to hemoglobin, so interfering with this carrier molecule limits oxygen delivery to the periphery. Hemoglobin increases the oxygen-carrying capacity of blood by about 40-fold,with the ability of hemoglobin to carry oxygen influenced by the partial pressure of oxygen in the environment, a relationship described in the oxygen hemoglobin dissociation curve. When the ability of hemoglobin to carry oxygen is interfered with, a hypoxic state can result.
Anemia
Hemoglobin plays a substantial role in carrying oxygen throughout the body and when it is deficient, anemia can result, causing 'Anaemic hypoxia' if tissue perfusion is decreased. Iron deficiency is the most common cause of anemia. As iron is used in the synthesis of hemoglobin, less hemoglobin will be synthesized when there is less iron, due to insufficient intake, or poor absorption.
Carbon monoxide poisoning andCyanide poisoning
Carbon monoxide competes with oxygen for binding sites on hemoglobin molecules. As carbon monoxide binds with hemoglobin hundreds of times tighter than oxygen, it can prevent the carriage of oxygen.Histotoxic hypoxia results when the quantity of oxygen reaching the cells is normal, but the cells are unable to use the oxygen effectively, due to disabled oxidative phosphorylation enzymes. This may occur in Cyanide poisoning.
TYPES OF HYPOXIA –PHYSIOLOGICAL COMPENSATION
Biochemists usually define hypoxia as O2-limited electron transport .Physiologists and clinicians define hypoxia as a state of reduced O2availability or decreased O2 partial pressures below critical thresholds, thus restricting or even abolishing the function of organs, tissues, or cells. Anoxia describes the state where no O2is detected in the tissue (O2 partial pressure 0 mm of mercury [mmHg]).
Hypoxia can be categorized into two main types- Acute hypoxia and chronic hypoxia.These two subtypes can lead to completely different hypoxia-related responses within the tumor, which could have a direct effect on tumor development and response to treatment. In order to accurately assess the specific biological consequences, it is important to understand which time frames best define acute and chronic hypoxia.
Acute hypoxia
It is also called as perfusion limited hypoxia. It is caused by inadequate blood flow in tissues. Because of structural and functional abnormalities in tumor micro vasculatures such as disorganized vascular network, dilations, an incomplete endothelial lining etc. It leads to ischemic hypoxia.
Chronic hypoxia
It is called as diffusion limited hypoxia characterized by diffusion distances with tumor expansion. This leads to inadequate O2 supply for cells distant from nutrition blood vessels. Chronically hypoxic cells are usually situated remotely from capillaries.
Figure 1The vascular network of normal tissue versus tumor tissue[3]
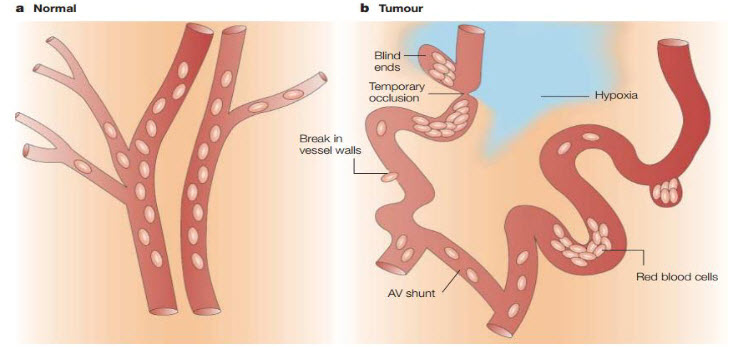
Tumors contain regions of hypoxia and necrosis because their vasculature can’t supply oxygen and other vital nutrients to all the cells. Whereas normal vasculature (a) is hierarchically organized, with vessels that are sufficiently close to ensure adequate nutrient and oxygen supply to all cells, tumor vessels (b) are chaotic, dilated, tortuous and are often far apart and have sluggish blood flow. As a consequence, areas of hypoxia and necrosis often develop distant from blood vessels. In addition to these regions of chronic (or diffusion-limited) hypoxia, areas of acute (or perfusion-limited) hypoxia can develop in tumors as a result of the temporary closure or reduced flow in certain vessels[4].
HYPOXIC TUMORS MICROENVIRONMENT – BIOLOGICAL ASPECTS
Hypoxia is property of solid tumors.Robust tumor growth requires the presence of a local vascular network that supplies both oxygen and nutrients to tumor cells.
Figure 2 Tumor growth[3]
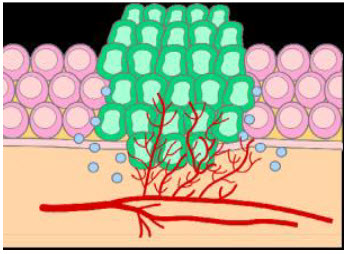
The basic difference between normal and tumor tissues is that they lack vasculature.
Figure 3 Normal vs. tumor tissues[3]
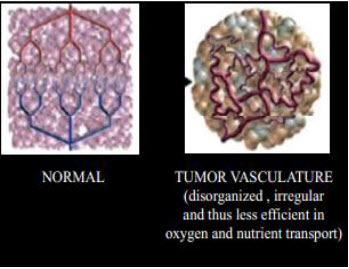
Properties of solid tumors:
1. High Heterogeneity
2. Low oxygen tension
3. Low pH
4. Low glucose concentration
Oxygenation status in tumors
Oxygen levels are typically very heterogeneous, both among patients and within individual tumors. Following Table 1depicts critical O2partial pressures below which adequate metabolic functions in solid tumors (metabolic hypoxia) cannot be maintained.
Table 1. Oxygenation of tumors and the normal tissue[4]
|
Tumor type |
Tumor mmHg |
Normal mmHg |
|
Glioblastoma |
4.9 |
- |
|
Head and neck carcinoma |
14.7 |
45 |
|
Lung cancer |
7.5 |
39 |
|
Breast cancer |
10 |
- |
|
Pancreatic cancer |
2.7 |
51.6 |
|
Cervical cancer |
5.0 |
51 |
|
Prostate cancer |
2.4 |
30 |
|
Soft-tissue sarcoma |
6.2 |
- |
During the past decade, the oxygenation status of solid tumors has been evaluated by investigators in many specialized centers. Despite various limitations of the techniques used, a number of key findings have been described as follows:
1. Most tumors have lower median O2partial pressures than their tissue of origin;
2. Solid tumors contain areas of low O2 partial pressure that cannot be predicted by clinical size, stage, grade, histology, and site;
3. Tumor-to-tumor variability in oxygenation is usually greater than intratumor variability in oxygenation;
4. Recurring tumors have a poorer oxygenation status than the corresponding primary tumors.
The oxygenation status has been measured using following techniques[5]:
1) Invasive micro sensor techniques for direct tissue measurements -
Polarographic sensors
Luminescence-based optical sensors
2) Electron paramagnetic resonance oximetry
3) Techniques for intravascular detection
Cryospectrophotometry
Near-infrared spectroscopy
Phosphorescence imaging
4) Nuclear magnetic resonance spectroscopy and imaging techniques -
1H-MRI
19F-magnetic resonance relaxometry
5) Noninvasive detection of sensitizer adducts
[18F]Fluoromisonidazole
[123I]Iodoazomycin-arabinoside
6) Invasive immunohistochemical hypoxia marker techniques –
Misonidazole[3H-labeled]
Pimonidazole
Etanidazole
Nitroimidazole-theophylline
The present “gold standard” is intratumor polarographic measurement of O2 partial pressures by using microsensor techniques that adhere to the systematic random sampling principle.However, no single method will probably be suitable for all situations; therefore, where possible, use of more than one technique may be advisable. In all instances, careful interpretation of the data obtained is paramount, and researchers should bear in mind the exact parameters measured and the limitations of the specific methods used[5].
Hypoxia mediated metabolic changes
When an unrestricted supply of oxygen is available, for most tumors, the rate of O2consumption (respiration rate) and adenosine triphosphate (ATP) production is comparable to that found in the corresponding normal tissue, despite the deregulated organization of cells in malignant tumors. To maintain a sufficient energy supply for membrane transport systems and synthesis of chemical compounds, an adequate supply of O2is required.
Warburg effect[6]:
In oncology, the Warburg effect is the observation that most cancer cells predominantly produce energy by a high rate of glycolysis followed by lactic acid fermentation in the cytosol, rather than by a comparatively low rate of glycolysis followed by oxidation of pyruvate in mitochondria as in most normal cells. Malignant, rapidly growing tumor cells typically have glycolytic rates up to 200 times higher than those of their normal tissues of origin.
Figure 4 Warburg effect[6]
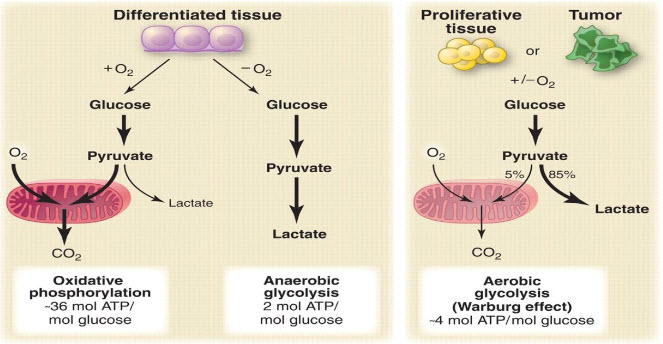
A particular change in metabolism, known as the Warburg effect results in high rates of glycolysis in both normoxic and hypoxic cancer cells. Generally normal cells use TCA cycle which is a series of chemical reactions used by all aerobic organisms to generate energy through the oxidation of acetate derived from carbohydrates, fats and proteins into carbon dioxide and chemical energy in the form of adenosine triphosphate (ATP) but in hypoxia tumor cells use glycolysis which isthe metabolic pathway that converts glucoseinto pyruvate, the free energy released in this process is used to form the high-energy compounds ATP (adenosine triphosphate) and NADH (reduced nicotinamide adenine dinucleotide).
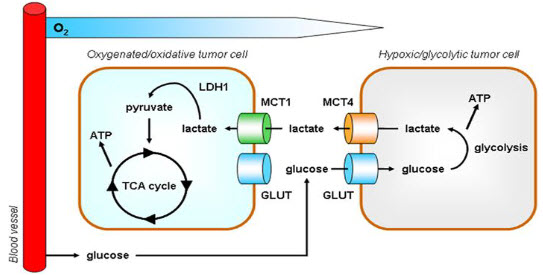
Figure 5 Hypoxia – TCA cycle to Glycolysis[6]
Changes in pH
In the glycolysis metabolic pathway glucose is converted into pyruvate which further converted into lactate. In the hypoxic conditions , lactate concentration increases very often so it is observed that hypoxic tumor is associated with low pH. It is proved that pH is substantially lower in hypoxic tissues than normal tissues by electrode method of analysis.
CELLULAR RESPONSES TO HYPOXIA
Hypoxia can influence tumor cells in one of two ways either by acting as a stressor that impairs growth or causescell death (slowing of proliferation, apoptosis, or necrosis)or by serving as a factor that ultimately results in malignantprogression and increased resistance to radiation therapyand other cancer treatments. It increases in malignant progression and treatment resistanceare manifestations of hypoxia-induced proteomic andgenomic changes within the tumor cells[7].
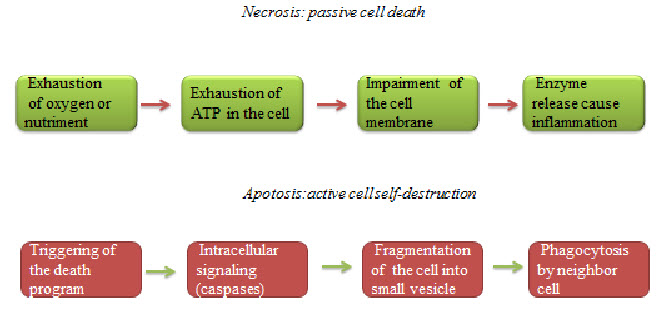
Hypoxic cells death can be seen by any of the two ways necrosis and apoptosis.Necrosis is a form of cell injury that results in the premature death of cells in living tissue by autolysis. Apoptosis is a naturally occurring programmed and targeted cause of cellular death.
Figure 6 Necrosis and apoptosis[7]
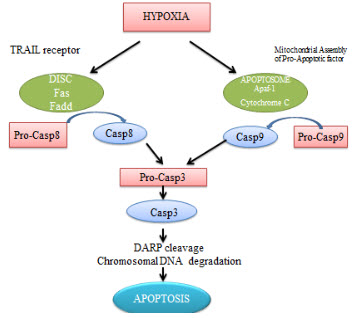
Apoptosis
It is proved that cell death pathway for the hypoxic cells are apoptosis. Under hypoxic conditions, caspase-3 gets activated in many cells like endothelial cells.Literature survey suggests that oxygen deprivationinduced apoptosis is transcriptionally regulated by the transcription factor p53.
Figure 7 Apoptosis in hypoxia[7]
HYPOXIA - GENE EXPRESSION HIF-1
Cells that are poorly oxygenated (<7 mmHg) display a series of adaptive responses that allow for survivaland continued proliferation.Tumor hypoxia negatively regulates cell growth andcauses a more malignant phenotype by increasing theexpression of genes encoding angiogenic, metabolic andmetastatic factors.Thehypoxia-inducible factor (HIF)-1 is a master transcriptional activator of oxygen-regulated genes.Hypoxia-inducible factors (HIFs) are transcription factors that respond to changes in available oxygen in the cellular environment.
HIF -1
HIF-1 is a heterodimeric basic HLH-PAS protein that consists of αand βsubunits.HIF-1 belongs to the PER-ARNT-SIM (PAS) subfamily of the basic helix-loop-helix (bHLH) family of transcription factors.The alpha and beta subunit are similar in structure and both contain the following domains[9].
- N-terminus – a bHLH domain for DNA binding
- Central region-Per-ARNT-Sim(PAS)domain, which facilitates heterodimerization
- C-terminus – recruits transcriptional coregulatory proteins
Figure 8 HIF -1[9]
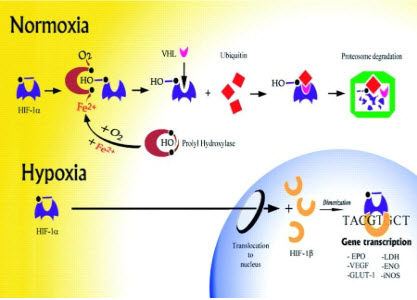
HIF-1β(ARNT) is constitutively expressed, HIF-1αis precisely regulated by cellularoxygen levels. Under hypoxic conditions, HIF-1αis induced, dimerizes with a βsubunit, translocates to the nucleus and initiates gene transcription.
Figure 9 Regulation of HIF -1[9]
The HIF signaling cascade mediates the effects of hypoxia, the state of low oxygen concentration, on the cell. Hypoxia often keeps cells from differentiating. However, hypoxia promotes the formation of blood vessels, and is important for the formation of a vascular system in embryos, and cancer tumors.
HIF-1 induced changes in gene expression
It is seen that under hypoxic conditions around 40 genes have been expressed by the HIF-1 factor.
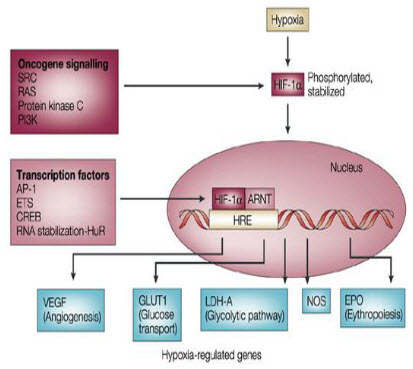
Figure 10 Hypoxia induced genes[9]
GLUT1 transporter expression
GLUT 1is a member of the GLUT transporter family of 14 hexose transporters responsible for facilitating the transport of hexose sugars along the concentration gradient. GLUT1 is the most abundantly expressed of the family thought to maintain basal glucose transport in almost all cell types. GLUT1 levels, in response to hypoxic conditions, have been shown to increase with changes at both the mRNA and protein levels.Moreover, transport of GLUT1 has been shown to increase under these hypoxic conditions. With the role of transporting sugars from the extracellular to the intracellular environment, GLUT1, along with other members of the GLUT family, can be rate-controlling for cellular glycolytic metabolism. Having an increased level of GLUT1, in the case of hypoxic tumors, increases the flux of glucose into the cells allowing for a higher rate of glycolysis and thus greater risks of metastasis.
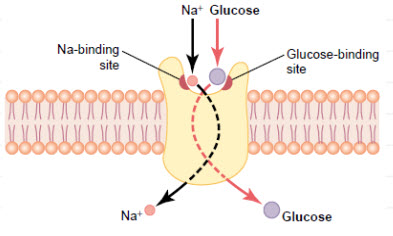
Figure 11 GLUT1 Glucose Transporters[10]
Basically HIF-1 activates the transcription of glycolytic enzymes such as aldolases A and C, enolase1,hexokinases1and3, lactate dehydrogenase A, phosphofructokinase L and phosphoglycerate kinase (PGK) 1.
Regulation of the growth of new blood vessels (angiogenesis) in primary and metastatic tumors is very important for malignant progression. Among the various factors that regulate angiogenesis, vascular endothelial growth factor (VEGF) is known to be strongly up regulated by hypoxia. VEGF expression is increased in regions near necrosis in tumors and multicell spheroids in vitro. Studies are ongoing in a number of research groups to assess the relationships between hypoxic regions, VEGF expression, and the development of new blood vessels. Such studies are especially important because of the apparent paradoxical results that both increased angiogenic index and increased hypoxia are predictive of poor prognosis. Hypoxia has also been shown to increase the transient induction of the anti-angiogenic gene, thrombospondin-1 (TSP-1) in cells containing a wild-type p53 gene .On the other hand, the basal and hypoxia-inducible expression of TSP-1 was undetectable in cells with mutant p53. In contrast, VEGF was induced under hypoxic conditionsregardless of the cellular p53 genotype. These studies suggest that tumor oxygenation status together with tumor cell genotype play and important role in angiogenesis.
Figure 12 Expression of HIF-1 in human cancer[11]
Regulation of HIF-1αexpression
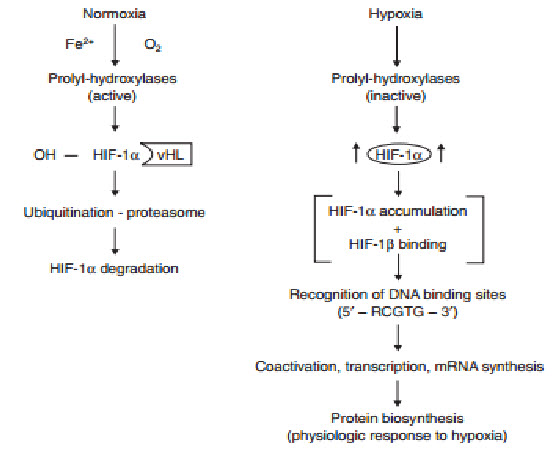
determines the subjection of HIF-1αto proteinhydroxylation. Under normoxic conditions, ubiquitination of HIF-1αtargets the subunit for proteasome degradation. Under hypoxic conditions, HIF-1βdimerizes withHIF-1α and the active HIF-1dimer binds to hypoxia response elements containing the corerecognition sequence 5′-RCGTC-3′ and then recruits coactivator molecules, resulting in the formation of an increased transcription initiation complex and mRNA synthesis,leading ultimately to the biosynthesis of proteins that mediate responses to hypoxia.
The oxygen-dependent regulation of HIF-1α occurs at the level of protein degradation.Under hypoxic conditions, stabilization of HIF-1αis negatively regulated by a series of oxygen-dependent post translational modifications, which are mediated by three prolyl hydroxylases and one or more asparaginyl hydroxylases. A conserved proline residue, proline564, is hydroxylated under normoxic conditions, allowing the von Hippel-Lindau (VHL) E3 ubiquitin ligase complex to bind to HIF-1α.The VHL complex adds ubiquitin to HIF-1α,which is then degraded by proteasomes. Under hypoxic conditions, oxygen becomes rate-limiting for prolyl hydroxylation,resulting in decreased ubiquitination of HIF-1α[10,11].
Figure 13 Regulation of HIF-1α[11]
HIF-1 independent pathways
Although HIF-1 seems to play a pivotal role in hypoxic response, other hypoxia-regulated transcription factors do exist. For example, NF-κB can also be activated by hypoxia. Activation of NF-κB leads to transcription of target genes such as those encoding proinflammatorycytokines (e.g., interleukins 6 and 8, TNF-α) and cyclooxygenase-2 (COX-2). COX-2 has angiogenic and growth-stimulatory properties, and is able to activate the genes for urokinase-like plasminogen activator and matrix metalloproteinase-2, both of which are associated with tumor invasiveness. NF-κB has also been shown to play an important role in apoptosis regulation since it leads to over expression of the antiapoptotic factor bcl-2.
AP-1 has also been identified as a hypoxia-inducible transcription factor. Prolonged AP-1 activation by hypoxiamay depend on HIF-1α, with both of these cooperating inthe transactivation of target genes[12,13].
Genomic changes
The tumor microenvironment is considered hostile, being characterized by areas of chronic or transient hypoxia, low pH, nutrient deprivation, and energy depletion. In a classic study, Reynoldsand colleagues examined the consequences of tumor growth under these conditions, using a tumorigenic cell line carrying a recoverable, chromosomally based lambda phage shuttle vector designed to identifymutations without the need for a genetic selection of mutant cells .The cells were grown concurrently either in culture or as tumors in nude mice. The frequency of mutations in the cells within the murine tumors was found to be five times that of the comparator cultured cells (9.3× versus1.8× , respectively;p < 0.0001). Moreover, the mutation patterns of the two cell groups differed, with the tumor grown cells displaying significantly more deletions and transversions than those grown in culture. Particularly noteworthy is the finding that exposure of cultured cells to hypoxic conditions produced an elevated mutation frequency and a mutation pattern similar to those observed in the tumor-grown cells. These findings suggest that the typeof genetic instability found in malignant tumors may in part be the consequence of specific mutagenic properties of the hypoxic micro environment.
Tumor cell survival and proliferation or, alternatively, growth impairment, stasis, and cell death are not solely dependent on proteomic changes. Mutations in oncogenes and tumor suppressor genes are generally thought to be of crucial importance for the development of tumor aggressiveness. Hypoxia ( ≤0.7 mmHg) promotes genomic instability, thereby increasing the number of mutations (genetic variants). Hypoxia concomitantly exerts a strong selection pressure. Tumor cell variants with adaptations favorable to survival under hypoxic conditions (e.g., lower capacity for cell-cycle arrest or apoptosis, greater angiogenic potential) may have growth advantages over non adapted cells in the hypoxic microenvironment and expand through clonal selection. The expansion of cell clones with favorable proteomic and genomic adaptive changes can, in turn, exacerbate tumor hypoxia, thereby establishing a vicious circle of increasing hypoxia and subsequent malignant progression. At the clinical level, the consequences of this vicious circle are translated intomore local recurrences, locoregional spread, and distant tumor metastases, and greater resistance to radiation therapy and certain forms of chemotherapy[14].
p53 and proteomic changes
Evidence from recent studies suggests that sustained (>6-8 hours) or fluctuating hypoxic stress (≤7 mmHg) can lead to alterations (stimulation or inhibition) of geneexpression, as well as posttranscriptional and posttranslational modulations that result in changes in the tumor cell proteome[15].These hypoxia-induced proteomic changes may, in turn, lead to growth stasis or impairment through molecularly mediated cell-cycle arrest, differentiation, programmed cell death (apoptosis), or necrosis . Hypoxia-induced cell-cycle arrest at the G1/S check point may be triggered by a hypoxia-inducible factor one alpha (HIF-1α)-mediated activation of the cyclin-dependent kinase inhibitors p21 and p27 . This response seems to be independent of p53, even though p53 accumulates underhypoxia . Instead, an increased level of p53 under hypoxic conditions may lead to the alternative activation of apoptosis with Apaf-1 and caspase-9 as downstream effectors .However, hypoxia may also induce p53-independent apoptosis pathways involving genes of the BCL-2 family and others. Experimental studieshave suggested that hypoxia may act as a morphogen to induce terminal differentiation of cells, and hypoxia is known to result in necrotic cell death. Overall, the effects of hypoxia-related proteomic changes leading to tumor cell growth stasis and death may explain the delayed recurrences, dormant micro metastases andgrowth retardation observed in large tumor masses. Alternatively, hypoxia-induced proteomic changes may promote tumor propagation by enabling cells to adapt to nutritive deprivation, or by facilitating proliferation, local invasion, and metastatic spread, there by permitting the cells to escape their hostile environment. One of the major promoters of tumor cell adaptation to hypoxic stress is the transcription factor HIF-1, which accumulates in response to declining cellular levels[15].HIF-1 activates a battery of more than 30 genes, many of which express protein products involved in delivery (e.g., erythropoietin), angiogenesis (e.g., vascular endothelial growth factor [VEGF]), energy preservation (e.g., glucose transporters and glycolytic enzymes), and other processes essential to tumor cell survival, propagation, and spread . Angiogenesis is an especially important factor in tumor progression since a tumor usually cannot grow beyond ~1 mm in diameter without an adequate blood supply.
The transcription factor p53 lies at the center of a protein network that controls cell cycle progression and commitment toapoptosis. p53is inactive in proliferating cells, largely because of negative regulation by the Mdm2 oncoprotein, with which it physically associates. Release from this negative regulation is sufficient to activate p53 and can be triggered in cells by multiple stimuli through diverse pathways. This diversity is achieved in part because Mdm2 uses multiple mechanisms to inactivate p53: it targets p53 for ubiquitination and degradation by proteosomes, shuttles it out of the nucleus and into the cytoplasm, prevents its interaction with transcriptional coactivators, and possesses an intrinsic transcriptional repressor activity.Homozygous deletion of p53 promotes neovascularizationand growth of tumor xenografts in nude mice. Because p53 promotes Mdm2-mediated ubiquitination and proteasomal degradation of HIF-1α, loss of p53 in tumor cells enhances HIF-1α levels and augments HIF-1-dependent transcriptional activation
of the VEGFgene, which is a master gene for malignant progression in response to hypoxia[16].
TARGETED CANCER THERAPY - HYPOXIA
The compelling evidence for hypoxia in tumor tissue and its therapeutic importance makes hypoxia a high priority target for cancer therapy.A characteristic feature of solid tumors is the presence of cells at very low oxygen tensions. These hypoxic cells confer radiotherapy and chemotherapy resistance to the tumors, as well as selecting for a more malignant phenotype.These hypoxic cells, however, provide a tumor-specific targeting strategy for therapy, and four approaches are beinginvestigated: prodrugs activated by hypoxia; hypoxia-selective gene therapy; targeting the hypoxia-inducible factor 1(HIF-1) transcription factor; and the use of recombinant obligate anaerobic bacteria[17,18].
Bioreductive prodrugs
The concept of activating prodrugs selectively in tumors, to achieve targeted delivery of cytotoxins, has a long history. The first clear demonstration was the reactivation of β-glucuronide metabolites of an aniline nitrogen mustard in tumors with high β-glucuronidase activity, but such approaches have struggled with the challenge of finding tumors with high enough expression of the activating enzymes to achieve useful selectivity. Hypoxia is potentially a more generic feature, with a clear basis for tumor selectivity, although expression of the activating enzymes is also critically important in this context.Five different chemical moieties (nitro groups, quinones, aromatic N-oxides, aliphatic N-oxides and transition metals) have the potential to be metabolized byenzymatic reduction under hypoxic conditions, and thus provide the basis for the design of bioreductive prodrugs for exploiting tumor hypoxia. Most often these mechanisms involve the re-oxidation by oxygen of the initial free radical intermediate formed by a one-electron reduction of the prodrug, thus generating superoxide. This futile redox cycling ensures that steady-state concentrations of the prodrug radical are kept low in oxic cells, resulting in hypoxia-selective cell killing provided that the prodrug radical (or its downstream products) is more cytotoxic than superoxide or the unreduced prodrug[19].
Inhibition of drug reduction by oxygen through this redox cycling mechanism was first demonstrated for nitro compounds and was subsequently shown to be responsible for the hypoxia-selective cytotoxicity of nitroimidazoles. This bioreductive mechanism is distinct from hypoxic cell radiosensitization by the same compounds, which is due to the ability of these compounds to replace oxygen in oxidizing ionizing radiation-induced DNA free radicals to generate cytotoxic DNA strand breaks.This first proof-of-principle demonstration of the hypoxia-selective cytotoxicity of bioreductiveprodrugactivity stimulated the search for ways of linking nitroreduction to the formation of more potent cytotoxins and for other redox moieties capable of hypoxia-selective metabolic activation
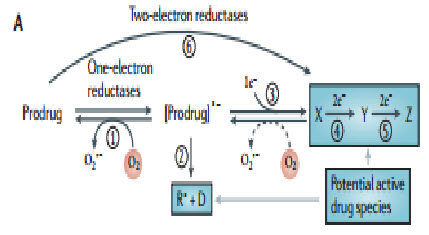
Figure 14 Prodrug activation[19]
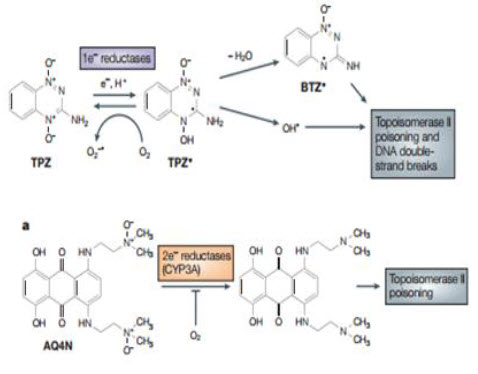
Bioreductive drugs are compounds that are reduced by biological enzymes to toxic, active metabolites.They are designedsuch that this metabolism occurs only or preferentially in the absence of oxygen. Tirapazamine (TPZ) is the leading compound in this class of agents. Under hypoxic conditions, TPZ is reduced to a radical that leads to DNA double-strand breaks (DSBs), single-strand breaks, and base damage. Recently, TPZ has been shown to be a hypoxia-selective topoisomerase II poison.Another promising bioreductive drug is AQ4N, a prodrug that is activated by reduction in hypoxic cells, producing a stable product (AQ4) that intercalates with DNA and blocks topoisomerase II action. AQ4N is not effective when given alone, but shows substantial antitumor activity when combined with methods to increase the hypoxic fraction, with radiation or with other anticancer drugs[20].
Figure 15 Mechanism of action[20]
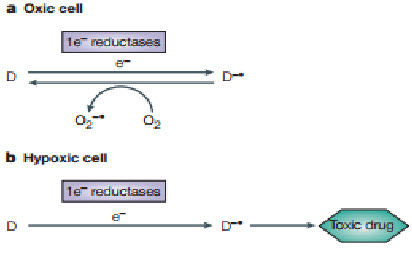
A common mechanism by which a non-toxic prodrug can be activated to a toxic drug in a hypoxia-dependent manner is explained above.In essence hypoxia-selective cytotoxicity requires one-electron reduction of a relatively non-toxic prodrug to a radical that then becomes a substrate for back-oxidation by oxygen to the original compound. If the so-formed radical or downstream products of the radical are much more toxic than the superoxide generated by redox cycling in oxic cells, hypoxia-dependent cytotoxicity arises. Examples of hypoxia-activated prodrugs in clinical trials are illustrated below[21].
Tirapazamine Brown and Lee discovered the hypoxic cytotoxicity of tirapazamine (TP2) almost 20 years ago, and this is the first compound tobe developed specifically as a hypoxic cytotoxin and for which antitumor activity has been demonstrated in clinical trials. Before the discovery of this benzotriazine di-N-oxide, two other classes of agents were known that produced some selective killing of hypoxic cells: quinone-containing alkylating agents, of which mitomycin C is the prototype; and nitro aromatic compounds, of which misonidazole and RB 6145 areexamples. TPZ was a significant advance over the previously known classes because its differential toxicity towards hypoxic cells was larger and combination studies with fractionated radiation demonstrated itsability to kill hypoxic cells in transplanted tumor The mechanism for selective toxicity of TPZ tohypoxic cells follows the general scheme outlined below.However whereas it had previously been proposed thatthe damaging species was the TPZ radical itself ,it now seems that the toxic species is an oxidizing radical formed by spontaneous decay of the protonated TPZ radical; this ultimate cytotoxin has been indicated to be either the hydroxyl radical or a benzotriazinyl (BTZ) radical formed by loss of water The oxidizing radical gives rise to cytotoxic DNA double-strand breaks through a TOPOISOMERASE-II-dependent process. TPZ potentiates the antitumor effect of radiation by selectively killing the hypoxic cells in the tumors. As these are the most radiation-resistant cells in tumors, TPZ and radiation act as complementary cytotoxins, each one killing the cells resistant to the other, thereby potentiating the efficacy of radiation on the tumor. TPZ is also very effective in enhancing the anticancer activity of the chemotherapeutic drug cisplatin, an interaction that again depends on hypoxia but that results from an increase in cisplatinsensitivity in non-lethally-damaged TPZ-treated cells rather than from complementary killing of oxic and hypoxic cells by the two agents, as is the case with radiation. The interaction with cisplatin has been tested in a Phase III clinical trial with advanced non-smallcell lung cancerand has been shown to be effective - the addition of TPZ to the standard cisplatinregimen doubled the overall response rate and significantly prolonged survival. TPZ has also been tested in a randomized Phase II trial with cisplatin-based chemoradiotherapy of advanced head and neck cancer and the preliminary results of this trial also show improved survival in the group treated with TPZ. A Phase III study with cisplatin-based chemoradiotherapy is now underway. Although TPZ seems to have clinical activity, and therefore provides important proof of principle for this approach, the dose that can be administered during chemoradiation is limited by neutropaenia and other toxicities by as yet unknown mechanisms. So, there is a clear need for improved hypoxia-activated prodrugs[22,23].
AQ4N The only other hypoxia-activated prodrug now in clinical trials - the anthraquinone AQ4N was designed specifically for this purpose. It resembles TPZ in being a di-N-oxide, but has a distinct mechanism of activation and cytotoxicity. AQ4N is a prodrug of a potent DNA intercalator/topoisomerase poison, AQ4, which is formed by reduction of the two tertiary amine N-oxide groups that mask DNA binding in the prodrug form.AQ4N is unusual among hypoxia-activated prodrugs in being activated by two-electron reduction, which is effected mainly by the CYP3A members of the cytochrome P450 family, which are strongly expressed in some human tumors.Inhibition by oxygen results from competition between and prodrug for binding at the reduced haem group in the enzyme active site, rather than from redox cycling.Although AQ4 is selective for cycling cells, its long residence time in tissue probably enables it to persist until hypoxic cells come into cycle. AQ4N has substantial activity against hypoxic cells in various transplanted tumors and has recently completed a Phase I clinical trial. The results of further clinical evaluation are awaited with interest.
Figure 16 TPZ and AQ4N mechanism[23]
Gene therapy
A key limitation of present day gene therapy of cancer is the lack of specificity of the gene-delivery system. Accordingly, essentially all of the protocols now being investigated in cancer gene therapy involve local administration of the delivery vectors directly into the tumor, usually by needle injection. Although this might be useful in some cases, it has limited applicability to cancer in general because metastases from the primary tumor are usually too numerous, inaccessible or undetected to allow for direct injection. An alternative to direct targeting of tumors is to have the therapeutic gene transcribed or translated by a tumor-specific property so thatmacrophages are often recruited to tumors and that such macrophages show increased levels of HIF-1α in various human tumorsTo achieve high specificity to hypoxic tumor cells, many hypoxia-response promoters have been constructed to express therapeutic genes. Most of the known hypoxically-inducible genes involve a cis-acting HRE, which can be present in either the 5′or 3′flanking DNA regions. HREs contain one or more binding sites for either HIF-1αor other related basic helixloop-helix PAS (bHLH-PAS) proteins. Several hypoxia-inducible promoters have been constructed using HREs from hypoxia-responsive genes, such as phosphoglycerate kinase 1 (PGK-1), enolase, LDH-A, EPOand VEGF[24].
Following figure shows how the hypoxic environment of tumors, which produces high levels of the hypoxia-inducible factor 1 (HIF-1) transcription factor, could be used in gene therapy to produce tumor-specific expression of an enzyme that can metabolize a non-toxic prodrug into a toxic drug selectively in the tumor. A) In oxygenated tissue, there is little or no HIF-1 transcription factor. Also, oxygen inhibits HIF-1 transactivation. Consequently, no prodrug-metabolizingenzyme is produced, so little or none of the prodrug isconverted to the toxic drug. B) In hypoxic tumor tissue, HIF-1 is produced and downstream genes are transcribedfollowing binding of HIF-1 to the hypoxia-responsive elements (HREs) in the promoter region of the genes. Therefore, the prodrug-activating enzyme with HREs in its promoter will be transcribed and, after translation, activate the prodrug to the toxic drug selectively in the tumor.
Figure 17 Gene therapy[24]
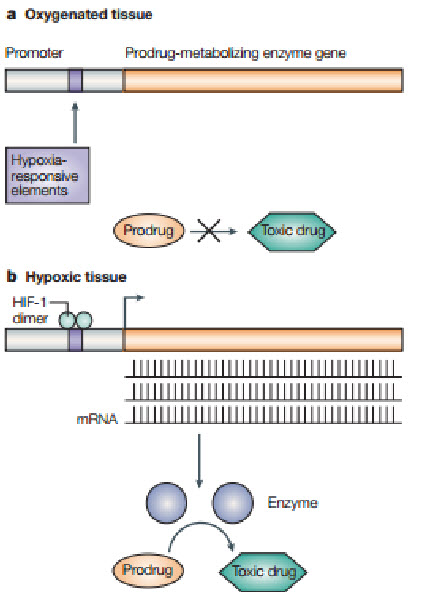
Targeting HIF-1
The characteristics, functions and possibilities for targeting HIF-1 in cancer therapy have been recently reviewed.Its role in angiogenesis, glucose utilization and tumor-cell survival ,its association with poor prognosis,and the fact that growth of mouse xenografts is inhibited by loss of HIF-1 activity all make it a potentially attractive tumor-specific target. It should be noted that the expression of HIF-1 is not restricted to hypoxic cells alone in many tumors, but is also upregulated by oncogenic mutations in RAS, SRC or ERBB2(also known as HER2/NEU). Therefore, targeting HIF-1 could potentially target the better oxygenated cells in the tumors.Three general approaches could be used to exploit the high levels of HIF-1αin cancers[25].
First, inhibition of transactivation of HIF-1 target genes (such as the angiogenesis inducer vascular endothelial growth factor) would be expected to have an antitumor effect. Proof of principle of this approach comes from studies by Kung and colleagues, who showed that tumor cells infected with a polypeptide that disrupted the binding of HIF-1α to its transcriptional coactivators p300/CREB, thereby inhibiting hypoxia-induced transcription, markedly reduced the growth of these cells whentransplanted into nude mice.These data have ledinvestigators to screen for small molecules thatinhibit HIF-1 transcription, and early reports have indicated that such compounds exist.However, anticancer effects directly attributable to inhibition of HIF-1 transactivation have yet to be reported. A second approach is to suppress HIF-1 protein levels, either by destabilizing the protein or inhibiting its production. The heat-shock protein 90 inhibitor geldanamycinhas been shown to reduce HIF-1 protein levels by promoting its oxygen and VHL independent degradation through the proteasome. However, it has yet to be demonstrated that this occurs in vivo or that the antitumor activity of this compound is a direct result of reduced levels of HIF-1, as many other proteins are also affected. Targeting of HIF-1 by direct injection of an antisense construct to HIF-1αhas been shown to eradicate a small transplanted thymic lymphoma and to increase the efficacy of immunotherapy against larger tumor. However, small-molecule inhibitors of HIF-1 would be preferable, and two groups have reported success. Mabjeesh and colleagues reported that microtubule inhibitors such as 2-methoxyestradiol, vincristine and paclitaxel reduce HIF-1αlevels in vitro apparently by inhibiting translation of HIF- 1αmRNA90. These compounds can also reduce tumor growth and vascularity, but whether this is an effect of reduced levels of HIF-1 or a direct effect on microtubules is not known. The second small molecule that has been reported to reduce HIF-1αlevels and inhibit tumour growth is the soluble guanylyl cyclase stimulator YC-1 Soluble guanylyl cyclase is the receptor for nitric oxide (NO) — a molecule involved in many signaling pathways, including those regulating vascular tone and platelet function. However, the authors attribute the antitumour and anti-angiogenic effects of YC-1 to a reduction in HIF-1αprotein levels (by an unknown post-translational effect) rather than to an effect on NO signaling. A third approach would be to screen for compounds that are preferentially toxic to cells expressing HIF-1α.At present this is a theoretical possibility with no published data demonstrating its efficacy [26]
Adenovirus:
Gene-Directed Enzyme Prodrug Therapy (GDEPT) involves the delivery to the target cells of a foreign gene encoding a non-toxic enzyme, which activates specific prodrugs to toxic agents at the site of conversion. Binley et al. constructed an optimized hypoxia response promoter (OBHRE) and put the herpes simplex virus thymidine kinase (HSVTK) gene under the control of the OBHRE. They then investigated hypoxia-targeted gene expression in vivo in the context of an adenovirus vector. Systemic administration of adenovirus followed by treatment with the anti-herpes viral agent ganciclovir (GCV), a HSV-TK substrate, resulted in tumor regression. Post and van Meir have developed a hypoxia/HIF-dependent replicative adenovirus (HYPR-Ad) that displayed hypoxia- dependent E1A expression and conditional cytolysis of hypoxic cells. Hernandez-Alcoceba et al. generated AdEHT2 and AdEHE2F, two conditionally replicative adenoviruses for the treatment of breast cancer. Minimal dual- specificity promoter that responds to estrogens and hypoxia controls the expression of the E1Agene in both oncolytic adenoviruses. The tumor selectivity of these adenoviruses is increased by introducing the telomerase reverse transcriptase promoter and the E2F-1 promoter, which are preferentially activated in cancer cells, into the E4 region of AdEHT2 and AdEHE2F, respectively [27].
Recombinant anaerobic bacteria:
Brown and colleagues first indicated that the necrotic regions in human solid tumors could be used to target cancer therapy to tumors using a genetically engineered non-pathogenic strain of the bacterial genus Clostridium. This genus comprises a large and heterogeneous group of Gram-positive, spore forming bacteria that become vegetative and grow only in the absence (or at very low levels) of oxygen. Malmgren and Flanagan were the first to demonstrate this phenomenon by observing that tumor bearing mice died of tetanus within 48 hours of intravenous injection of C. Tetani spores, whereas non-tumor-bearing animals were unaffected. Möse and Möselater reported that a nonpathogenic clostridial strain, C. butyricumM-55, localized and germinated in solid Ehrlich tumors in mice, causing extensive lysis without any concomitant effect on normal tissues. Such observations were soon confirmed and extended by several investigators using tumors in mice, rats, hamsters and rabbits, and were followed by clinical studies with patients with cancer. Although the anaerobic bacteria did not significantly alter tumor control or eradication, these clinical reports demonstrated that spores of nonpathogenic strains of clostridia could be given safely, that the spores germinate in the necrotic regions of tumors, and that lysis in these tumor regions can occur [30]. This is an important distinction over the similar approach using genetically modified, live attenuated Salmonella, which, although producing excellent colonization of transplanted tumors in mice, produced only marginal colonization of human tumors in a Phase I clinical trial. The reasons for the difference between the rodent and human tumors in colonization by Salmonella are unknown. However, colonization by clostridia is different from that of Salmonella in being dependent on hypoxic necrotic regions, which are equally common in human and rodent tumors. In addition, as noted above, excellent colonization of human tumors has been reported following intravenous injection of clostridial spores. The Clostridium used in the clinical studies was a strain of C. Sporogenes, renamed C. Oncolyticum to reflect the lysis that was produced in human tumours. This strain has been genetically modified to express the E.coli enzyme cytosine deaminase, which can convert the non-toxic 5-fluorocytosine to the toxic anticancer drug 5-fluorouracil. Animal experiments have demonstrated the efficacy of this approach and clinical studies are planned. In addition, other enzyme–prodrug systems for arming clostridia are in development, including CB 1954 (BOX 2), which, when activated by E.coli nitroreductase, kills non-cycling cells efficiently and is therefore expected to have greater activity against cells in hypoxic regions. Although clostridial-dependent enzyme prodrug therapy (CDEPT) is similar to the strategy of antibody-dependent enzyme prodrug therapy (ADEPT) which is now under clinical evaluation, it has several significant advantages, not the least of which is its favorable intratumour distribution. Because the prodrug-activating enzyme from clostridia will be at its highest concentration in areas adjacent to necrosis and far from blood vessels (FIG. 6), this guarantees the highest active drug concentrations in the distant cells and also minimizes the problem of leakage of activated drug back into the blood vessels, which has been reported to be a problem for ADEP[27]. A hypoxia-targeting fusion protein: To address the requirements for hypoxia- targeted therapy, namely high delivery efficiency, high specificity and selective cytotoxity, we have constructed a unique fusion protein, TOP3 [27].
High delivery efficiency: A severe limitation of protein therapeutics is the problem of transporting large proteins into cells. Overcoming this problem of bioavailability would not only enhance the effectiveness of existing therapeutics, but would also broaden the scope of viable cancer therapeutic strategies. The protein transduction domain embedded in the human immunodeficiency virus TAT protein (TAT-PTD); amino acids has been shown to successfully mediate the introduction of given peptides and proteins with a molecular weight in excess of 100,000 into mammalian cells in vitro and in vivo. TAT- PTD fusion proteins can be delivered to the entire body, including to the brain. Furthermore, delivery does not occur only via the bloodstream. TAT-PTD fusion proteins are also able to freely diffuse through cell membranes, and therefore through layers of tumor cells. Hence, they can be delivered to tissues where blood vessels are shut down, such as in the ischemic brain.
High specificity: As described above, HIF- 1αprotein stability is tightly regulated by oxygen-dependent prolyl hydroxylation and ubiquitination through its oxygen-dependent degradation domain (ODD), which contains proline 564. In vitro experiments have shown that endogenous HIF-1α is degraded within 5 min after exposure of cultured cells to 21%We have identified the minimum region of ODD (18–50 amino acids) that provides stability to a fusion protein under the oxygen-dependent conditions that degrade HIF-1α. The reporter protein TAT-PTD-ODD-β-galactosidase was examined for delivery efficiency and specificity. Systemic administration of TAT-PTD-ODD-β- galactosidase showed that it was successfully delivered and stabilized in hypoxic tumor cells, but not in normal liver cells. Therefore, if a cytotoxic protein is fused to the ODD polypeptide, its cytotoxicity will be controlled by the oxygen concentration.
Selective cytotoxity:
To obtain selective cytotoxity, we chose an endogenous cytotoxic protein, procaspase-3, which is a major executioner protease that is located at the most downstream position in several apoptotic pathways and remains dormant until initiator caspases activate it by direct proteolysis. It is activated specifically in hypoxic tumor cells because, as described above, the apoptotic pathway and, hence, initiator caspases are activated in hypoxic tumor cells, generating active caspase-3. The final fusion protein product TAT- PTD- ODD- Procaspase-3 (TOP3) (Fig. 18) was examined for its efficacy in tumor-bearing mice. Systemic administration of TOP3 significantly suppressed tumor growth and even reduced the tumor size without any apparent side effects in mice bearing human pancreatic tumor xenografts.
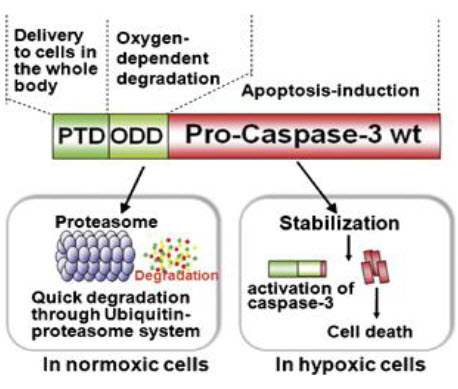
Figure 18 TOP3[27]
CONCLUSIONS
As the mechanisms underlying response to hypoxic stress are dissected at the molecular level in solid tumors, the existence of such a microenvironment in tumor cells is becoming apparent as a flaw of current cancer therapies. The difference between hypoxic tumor cells and well-oxygenated normal cells is so distinct that it is possible to design efficient and selective therapies against the sequestered regions. Whether targeting hypoxic tumor cells has a real benefit in clinical cancer therapy still remains an open question, however, since complete obliteration of hypoxic tumor cells has still not been achieved genetically or pharmacologically, even in in vivo experimental settings. Accumulation of data showing the impact of emerging hypoxia-targeting therapies, such as TOP3, on solid tumors should confirm tumor hypoxia as a major cause of malignant and refractory conversion. Such evidence would validate the idea of attacking tumor hypoxia as a therapeutic approach, and would provide momentum for further exploitation of this approach in clinical applications. Perhaps the most crucial requirement for hypoxia targeting strategies is the development of improved predictive tools for patient stratification. These tools need to evaluate not only hypoxia, but also many other determinants of sensitivity, as discussed above. Ultimately, tumor and host genomic analyses will revolutionize the matching of hypoxia-targeted therapeutics to individual patients. However, extracting information on physiological features such as the severity of hypoxia from genomic data will be challenging, so functional assays such as PET imaging are likely to play a major part in the foreseeable future. Together, this individualized phenotyping has the potential to identify clinical niches for the diverse types of cytotoxins that are already identified as hypoxia-selective, and provide a rational basis for their clinical development.
|
PharmaTutor (ISSN: 2347 - 7881) Volume 2, Issue 7 Received On: 27/04/2014; Accepted On: 09/05/2014; Published On: 01/07/2014 How to cite this article:PD Patil, R Tayade; A Potent Target for Cancer Treatment – Tumor Hypoxia Necrosis: An Overview; PharmaTutor; 2014; 2(7); 8-28 |
REFERENCES:
1. Brown, J. M.; Wilson, W. R. Exploiting Tumour Hypoxia in Cancer Treatment. Nat. Rev. Cancer 2004, 4, 437–447.
2. Chao, K. S.; Bosch, W. R.; Mutic, S.; Lewis, J. S.; Dehdashti, F.; Mintun, M. a; Dempsey, J. F.; Perez, C. a; Purdy, J. a; Welch, M. J. A Novel Approach to Overcome Hypoxic Tumor Resistance: Cu-ATSM-Guided Intensity-Modulated Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2001, 49, 1171–1182.
3. DeYoung, M. P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L. W. Hypoxia Regulates TSC1/2-mTOR Signaling and Tumor Suppression through REDD1-Mediated 14-3-3 Shuttling. Genes Dev. 2008, 22, 239–251. 4
. Garayoa, M.; Mart?, A.; Lee, S.; An, W. G.; Neckers, L.; Trepel, J.; Montuenga, L. M.; Ryan, H.; Johnson, R.; Gassmann, M. Expression in Human Tumor Cell Lines during Oxygen Deprivation?: A Possible Promotion Mechanism of Carcinogenesis. 2014, 1, 848–862.
5. Kizaka-Kondoh, S.; Inoue, M.; Harada, H.; Hiraoka, M. Tumor Hypoxia: A Target for Selective Cancer Therapy. Cancer Sci. 2003, 94, 1021–1028.
6. Koong, a C.; Mehta, V. K.; Le, Q. T.; Fisher, G. a; Terris, D. J.; Brown, J. M.; Bastidas, a J.; Vierra, M. Pancreatic Tumors Show High Levels of Hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 919–922. 7. Lewis, C. E.; Pollard, J. W. Distinct Role of Macrophages in Different Tumor Microenvironments. Cancer Res. 2006, 66, 605–612.
8. Park, J. E.; Tan, H. Sen; Datta, A.; Lai, R. C.; Zhang, H.; Meng, W.; Lim, S. K.; Sze, S. K. Hypoxic Tumor Cell Modulates Its Microenvironment to Enhance Angiogenic and Metastatic Potential by Secretion of Proteins and Exosomes. Mol. Cell. Proteomics 2010, 9, 1085–1099.
9. Sutherland, R. M. Tumor Hypoxia and Gene Expression--Implications for Malignant Progression and Therapy.ActaOncol. 1998, 37, 567–574.
10. Vaupel, P. The Role of Hypoxia-Induced Factors in Tumor Progression. Oncologist 2004, 9 Suppl 5, 10–17.
11. Wilson, W. R.; Hay, M. P. Targeting Hypoxia in Cancer Therapy. Nat. Rev. Cancer 2011, 11, 393–410.
12. Garayoa, M.; Mart?, A.; Lee, S.; An, W. G.; Neckers, L.; Trepel, J.; Montuenga, L. M.; Ryan, H.; Johnson, R.; Gassmann, M. Expression in Human Tumor Cell Lines during Oxygen Deprivation?: A Possible Promotion Mechanism of Carcinogenesis. 2014, 1, 848–862.
13. Groshar, D.; McEwan, a J.; Parliament, M. B.; Urtasun, R. C.; Golberg, L. E.; Hoskinson, M.; Mercer, J. R.; Mannan, R. H.; Wiebe, L. I.; Chapman, J. D. Imaging Tumor Hypoxia and Tumor Perfusion. J. Nucl. Med. 1993, 34, 885–888.
14. Harrison, L. B. Impact of Tumor Hypoxia and Anemia on Radiation Therapy Outcomes. Oncologist 2002, 7, 492–508. 15. Kizaka-Kondoh, S.; Inoue, M.; Harada, H.; Hiraoka, M. Tumor Hypoxia: A Target for Selective Cancer Therapy. Cancer Sci. 2003, 94, 1021–1028. 16. Koong, a C.; Mehta, V. K.; Le, Q. T.; Fisher, G. a; Terris, D. J.; Brown, J. M.; Bastidas, a J.; Vierra, M. Pancreatic Tumors Show High Levels of Hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 919–922.
17. Lewis, C. E.; Pollard, J. W. Distinct Role of Macrophages in Different Tumor Microenvironments. Cancer Res. 2006, 66, 605–612.
18. Liao, D.; Corle, C.; Seagroves, T. N.; Liao, D.; Corle, C.; Seagroves, T. N.; Johnson, R. S. Hypoxia- Inducible Factor-1 Α Is a Key Regulator of Metastasis in a Transgenic Model of Cancer Initiation and Progression Transgenic Model of Cancer Initiation and Progression. 2007, 563–572.
19. Maxwell, P. H.; Dachs, G. U.; Gleadle, J. M.; Nicholls, L. G.; Harris, a L.; Stratford, I. J.; Hankinson, O.; Pugh, C. W.; Ratcliffe, P. J. Hypoxia-Inducible Factor-1 Modulates Gene Expression in Solid Tumors and Influences Both Angiogenesis and Tumor Growth. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 8104–8109.
20. Robey, I. F.; Lien, A. D.; Welsh, S. J.; Baggett, B. K.; Gillies, R. J. Hypoxia-Inducible Factor-1α and the Glycolytic Phenotype in Tumors. Neoplasia 2005, 7, 324–330.
21. Schwab, L. P.; Peacock, D. L.; Majumdar, D.; Ingels, J. F.; Jensen, L. C.; Smith, K. D.; Cushing, R. C.; Seagroves, T. N. Hypoxia-Inducible Factor 1α Promotes Primary Tumor Growth and Tumor-Initiating Cell Activity in Breast Cancer. Breast Cancer Res. 2012, 14, R6.
22. Shahrzad, S.; Bertrand, K.; Minhas, K.; Coomber, B. L. Induction of DNA Hypomethylation by Tumor Hypoxia. Epigenetics 2007, 2, 119–125. 23. Sutherland, R. M. Tumor Hypoxia and Gene Expression--Implications for Malignant Progression and Therapy. ActaOncol. 1998, 37, 567–574.
24.Unruh, A.; Ressel, A.; Mohamed, H. G.; Johnson, R. S.; Nadrowitz, R.; Richter, E.; Katschinski, D. M.; Wenger, R. H. The Hypoxia-Inducible Factor-1 Alpha Is a Negative Factor for Tumor Therapy. Oncogene 2003, 22, 3213–3220.
25.Vordermark, D.; Kraft, P.; Katzer, A.; Bölling, T.; Willner, J.; Flentje, M. Glucose Requirement for Hypoxic Accumulation of Hypoxia-Inducible Factor-1alpha (HIF-1alpha). Cancer Lett. 2005, 230, 122– 133.
26. Wilson, W. R.; Hay, M. P. Targeting Hypoxia in Cancer Therapy. Nat. Rev. Cancer 2011, 11, 393–410.
27. Zhdanov, A. V; Dmitriev, R. I.; Golubeva, A. V; Gavrilova, S. a; Papkovsky, D. B. Chronic Hypoxia Leads to a Glycolytic Phenotype and Suppressed HIF-2 Signaling in PC12 Cells. Biochim.Biophys.Acta 2013, 1830, 3553–3569.






