

ABOUT AUTHORS:
*Sambhara Gayatri Deepti1, Aruna Ragidi1, A.M.S.Sudhakar Babu1, P.Venkateswara Rao2
1Department of Pharmaceutics
2Department of Pharmaceutical Analysis
A.M.Reddy Memorial college of Pharmacy, Narasaraopet, Guntur Dist., Andhra Pradesh, India.
gayatri.dpt11@gmail.com
ABSTRACT:
Liposomes are result of self assembly of phospholipid in an aqueous media resulting in closed bilayered structures. Liposomes are one of unique drug delivery system which can be use in controlling and targeting drug delivery system. Liposomes are generally classified based upon structure, method of preparation, composition and application, conventional liposome, and specialty liposome. Liposomes are formulated and processed to differ in size, composition, charge and lamellarity, depending upon method of preparation either active loading technique or passive loading technique. The prepared liposomes are characterized for visual appearance, liposomal size distribution, lamillarity, liposome stability, entrapped volume and surface charges. Different marketed formulations are available in market for liposomes. The liposomes have many applications which increase its importance over other formulations.
REFERENCE ID: PHARMATUTOR-ART-2021
INTRODUCTION:
Liposomes are vesicular structures composed of a lipid bilayer. These vesicular structures can be used as a vehicle for administration of nutrients and drugs. They are composed of natural phospholipids, and may also contain mixed lipid chains with surfactant properties. A liposome design may employ surface ligands for attaching to unhealthy tissue (e.g., cancers). The major types of liposomes are the multilamellar vesicle (MLV), the small unilamellar vesicle (SUV), and the large unilamellar vesicle (LUV). Drugs of both category (hydrophilic/ lipophillic) are easily embedded in the liposomes and are manufactured by methods such as Reverse Phase Evaporation Method, Sonication Method, Freeze Dried Rehydration Method.
Advances in liposome research have been able to allow liposomes to avoid detection by the body's immune system, specifically, the cells of reticuloendothelial system (RES). These liposomes are known as "stealth liposomes", and are constructed with PEG (Polyethylene Glycol) studding the outside of the membrane. These targeting ligands could be monoclonal antibodies (making an immunoliposome), vitamins, or specific antigens. Targeted liposomes can target nearly any cell type in the body and deliver drugs that would naturally be systemically delivered. Naturally toxic drugs can be much less toxic if delivered only to diseased tissues. In many hard diseases (cancers, tumors, HIV) the drug can be easily and effective delivered by the means of liposomes.
A liposome is an artificially-prepared vesicle composed of a lipid bilayer. The liposome can be used as a vehicle for administration of nutrients andpharmaceutical drugs.[2] Liposomes can be prepared by disrupting biological membranes (such as by sonication).
Liposomes are often composed of phosphatidylcholine-enriched phospholipids and may also contain mixed lipid chains with surfactant properties such as egg phosphatidylethanolamine. A liposome design may employ surface ligands for attaching to unhealthy tissue.[1]
Liposomes should not be confused with micelles and reverse micelles compose of monolayers.[4]
Figure.1. Liposome for Drug Delivery
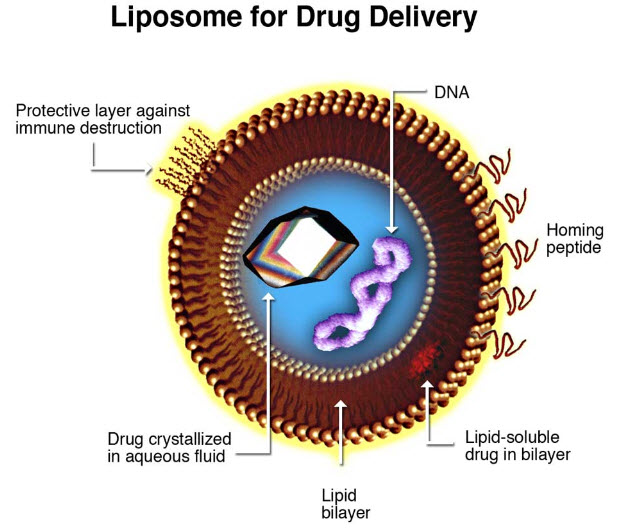
MECHANISM:
A liposome encapsulates a region of aqueous solution inside a hydrophobic membrane; dissolved hydrophilic solutes cannot readily pass through the lipids. Hydrophobic chemicals can be dissolved into the membrane, and in this way liposome can carry both hydrophobic molecules and hydrophilic molecules. To deliver the molecules to sites of action, the lipid bilayer can fuse with other bilayers such as the cell membrane, thus delivering the liposome contents. By making liposomes in a solution of DNA or drugs (which would normally be unable to diffuse through the membrane) they can be (indiscriminately) delivered past the lipid bilayer. A liposome does not necessarily have lipophobic contents, such as water, although it usually does.
Liposomes are used as models for artificial cells. Liposomes can also be designed to deliver drugs in other ways. Liposomes that contain low (or high) pH can be constructed such that dissolved aqueous drugs will be charged in solution (i.e., the pH is outside the drug's pI range). As the pH naturally neutralizes within the liposome (protons can pass through some membranes), the drug will also be neutralized, allowing it to freely pass through a membrane. These liposomes work to deliver drug by diffusion rather than by direct cell fusion.
A similar approach can be exploited in the biodetoxification of drugs by injecting empty liposomes with a transmembrane pH gradient. In this case the vesicles act as sinks to scavenge the drug in the blood circulation and prevent its toxic effect.[8] Another strategy for liposome drug delivery is to target endocytosis events. Liposomes can be made in a particular size range that makes them viable targets for natural macrophage phagocytosis. These liposomes may be digested while in the macrophage's phagosome, thus releasing its drug. Liposomes can also be decorated withopsonins and ligands to activate endocytosis in other cell types.
The use of liposomes for transformation or transfection of DNA into a host cell is known as lipofection.
FACTORS AFFECTING LIPOSOME PREPARATION:
The correct choice of liposome preparation method depends on the following parameters:
1. the physicochemical characteristics of the material to be entrapped and those of the liposomal ingredients;
2. the nature of the medium in which the lipid vesicles are dispersed
3. the effective concentration of the entrapped substance and its potential toxicity;
4. additional processes involved during application/delivery of the vesicles;
5. optimum size, polydispersity and shelf-life of the vesicles for the intended application; and,
6. batch-to-batch reproducibility and possibility of large-scale production of safe and efficient liposomal products
Formation of liposomes and nanoliposomes is not a spontaneous process. Lipid vesicles are formed when phospholipids such as lecithin are placed in water and consequently form one bilayer or a series of bilayers, each separated by water molecules, once enough energy is supplied.[19]
Liposomes can be created by sonicating phosphatidylcholine rich phospholipids in water. Low shear rates create multilamellar liposomes, which have many layers like an onion. Continued high-shear sonication tends to form smaller unilamellar liposomes. In this technique, the liposome contents are the same as the contents of the aqueous phase. Sonication is generally considered a "gross" method of preparation as it can damage the structure of the drug to be encapsulated. Newer methods such as extrusion and Mozafari method [20] are employed to produce materials for human use.
ADVANTAGES
Some of the advantages of liposome are as follows:
- Provides selective passive targeting to tumor tissues (Liposomal doxorubicin).
- Increased efficacy and therapeutic index.
- Increased stability via encapsulation.
- Reduction in toxicity of the encapsulated agents.
- Site avoidance effect.
- Improved pharmacokinetic effects (reduced elimination, increased circulation life times).
- Flexibility to couple with site specific ligands to achieve active targeting
DISADVANTAGES:
- low solubility
- short half life
- high production cost
- less stability
- leakage and fusion of encapsulated drug
- sometimes the phospholipid layer undergoes oxidation and hydrolysis reaction
STRUCTURAL COMPONENTS OF LIPOSOME :
There are number of the structural and nonstructural components of liposomes, major structural components of liposomes are:
a. Phospholipids :
Phospholipids are the major structural component of biological membranes, where two type of phospholipids exit- PHOSPHODIGLYCERIDES AND SPHINGOLIPIDS.
The most common phospholipid is phosphatidylcholine (PC) molecule. Molecule of phosphatidylcholine are not soluble in water and in aqueous media they align themselves closely in plannar bilayer sheets in order to minimize the unfavorable action between the bulk aqueous phase and long hydrocarbon fatty chain. The Glycerol containing phospholipids are most common used component of liposome formulation and represent greater than 50% of weight of lipid in biological membranes. These are derived from Phosphatidic acid.
Examples of phospholipids are:
1. Phosphatidyl choline (Lecithin) – PC
2. Phosphatidyl ethanolamine (cephalin) – PE
3. Phosphatidyl serine (PS)
4. Phosphatidyl inositol (PI)
5. Phosphatidyl Glycerol (PG)
b. Cholesterol :
Cholesterol dose not by itself form bilayer structure, but can be incorporated into phospholipid membranes in very high concentration upto 1:1 or even 2:1 molar ration of cholesterol to phosphatidylcholine. Cholesterol inserts into the membrane with its hydroxyl group oriented towards the aqueous surface and aliphatic chain aligent parallel to the acyl chains in the center of the bilayer. The high solubility of cholesterol in phospholipid liposome has been attributed to both hydrophobic and specific headgroup interation, but there is no unequivocal evidence for the arrangement of cholesterol in the bilayer.
CLASSIFICATION:
Liposomes have one or more bilayer structures.the vesicle size is an acute parameter in determining the size of liposome, and both size and number of bilayers determine the amount of drug encapsulated in the liposomes.
Liposomes are often distinguished according to their number of lamellae and size. Small unilamellar vesicles (SUV), large unilamellar vesicles (LUV) and large multilamellar vesicles (MLV) or multivesicular vesicles (MVV) are differentiated (see figure 2). SUVs show a diameter of 20 to approximately 100 nm. LUVs, MLVs, and MVVs range in size from a few hundred nanometers to several microns. The thickness of the membrane (phospholipid bilayer) measures approximately 5 to 6 nm.in the unilamellar liposomes the vesicle has a single phospholipid bilayer sphere enclosing the aqueous fluid.in the multilamellar liposome the vesicles have an onion structure.
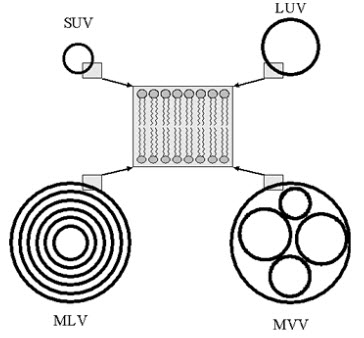
Figure 2. Schematic illustration of liposomes of different size and number of lamellae. SUV: Small unilamellar vesicles; LUV: Large unilamellar vesicles; MLV: Multilamellar vesicles;
PREPARATIONOF LIPOSOMES:
GENERAL METHOD OF PREPARATION AND DRUG LOADING:
Liposomes are manufactured in majority using various procedures in which the water soluble (hydrophilic) materials are entrapped by using aqueous solution of these materials as hydrating fluid or by the addition of drug/drug solution at some stage during manufacturing of the liposomes. The lipid soluble (lipophilic) materials are solubilized in the organic solution of the constitutive lipid and then evaporated to a dry drug containing lipid film followed by its hydration. These methods involve the loading of the entrapped agents before or during the manufacturing procedure (Passive loading). However, certain type of compounds with ionizable groups, and those which display both lipid and water solubility, can be introduced into the liposomes after the formation of intact vesicles (remote loading).
1. MECHANICAL DISPERSION METHODS:
Preparation of liposomes by lipid film hydration:
Preparation of Lipid for Hydration: When preparing liposomes with mixed lipid composition, the lipids must first be dissolved and mixed in an organic solvent to assure a homogeneousmixture of lipids. Usually this process is carried out using chloroform or chloroform:methanol mixtures. The intent is to obtain a clear lipid solution for complete mixing of lipids. Typically lipid solutions are prepared at 10-20mg lipid/ml of organic solvent, although higher concentrations may be used if the lipid solubility and mixing are acceptable. Once the lipids are thoroughly mixed in the organic solvent,the solvent is removed to yield a lipid film. For small volumes of organic solvent (<1mL), the solvent may be evaporated using a dry nitrogen or argon stream in a fume hood. For larger volumes, the organic solvent should be removed by rotary evaporation yielding a thin lipid film on the sides of a round bottom flask. The lipid film is thoroughly dried to remove residual organic solvent by placing the vial or flask on a vacuum pump overnight. If the use of chloroform is objectionable, an alternative is to dissolve the lipid(s) in tertiary butanol or cyclohexane. The lipid solutionis transferred to containers and frozen by placing the containers on a block of dry ice or swirling the container in a dry ice-acetone or alcohol (ethanol or methanol) bath. Care should be taken when using thebath procedure that the container can withstand sudden temperature changes without cracking. After freezing completely, the frozen lipid cake is placed on a vacuum pump and lyophilized until dry (1-3 days depending on volume). The thickness of the lipid cake should not be more than the diameter of the container being used for lyophilization. Dry lipid films or cakes can be removed from the vacuum pump, the container should be closed tightly and taped, and stored frozen until ready to hydrate
Hydration of Lipid Film/Cake:
Hydration of the dry lipid film/cake is accomplished simply by adding an aqueous medium to the container of dry lipid and agitating. The temperature of the hydrating medium should be above the gelliquid crystal transition temperature (Tc or Tm) ofthe lipid. After addition of the hydrating medium, the lipid suspension should be maintained above the Tc during the hydration period. For high transition lipids, this is easily accomplished by transferring the lipid suspension to a round bottom flask and placing the flask on a rotary evaporation system without a vacuum. Spinning the round bottom flask in the warm water bath maintained at a temperature above the Tc of the lipid suspension allows the lipid to hydrate in its fluid phase with adequate agitation.Hydration time may differ slightly among lipid species and structure, however, a hydration time of 1 hour with vigorous shaking, mixing, or stirring is highly recommended. It is also believed that allowing the vesicle suspension to stand overnight (aging) prior to downsizing makes the sizing process easier and improves the homogeneity of the size distribution. Aging is not recommended for high transition lipids as lipid hydrolysis increases with elevated temperatures. The hydration medium is generally determined by the application of the lipid vesicles. Suitable hydration media include distilled water, buffer solutions, saline, and non-electrolytes such as sugar solutions. Generally accepted solutions which meet these conditions are 0.9% saline, 5% dextrose and 10% sucrose. During hydration some lipids form complexes unique to their structure. Highly charged lipids have been observed to form a viscous gel when hydrated with low ionic strength solutions. The problem can be alleviated by addition of salt or by downsizing the lipid suspension. Poorly hydrating lipids such as phosphatidylethanolamine have a tendency to self aggregate upon hydration. Lipid vesicles containing more than 60 mol% phosphatidylethanolamine form particles having a small hydration layer surrounding the vesicle. As particles approach one another there is no hydration repulsion to repel the approaching particle and thetwo membranes fall into an energy well where they adhere and form aggregates. The aggregates settle out of solution as large flocculates which will disperse on agitation but reform upon sitting. The product of hydration is a large, multilamellar vesicle (LMV) analogous in structure to an onion, with eachlipid bilayer separated by a water layer. The spacing between lipid layers is dictated by composition with poly-hydrating layers being closer together than highly charged layers which separates on electrostatic repulsion. Once a stable, hydrated LMV suspension has been produced, the particles can be downsized by a variety of techniques, including sonication or extrusion.
Sizing of Lipid Suspension:
Sonication: Disruption of LMV suspensions using sonic energy (sonication) typically produces small,unilamellar vesicles (SUV) with diameters in the range of 15-50nm. The most common instrumentation for preparation of sonicated particles are bath and probe tip sonicators. Cup-horn sonicators, although less widely used, have successfully produced SUV. Probe tip sonicators deliver high energy input to the lipid suspension but suffer from overheating of the lipid suspension causing degradation. Sonication tips also tend to release titanium particles into the lipid suspension which must be removed by centrifugation prior to use. For these reasons, bath sonicators are the most widely used instrumentation for preparation of SUV.Sonication of an LMV dispersion is accomplished by placing a test tube containing the suspension in a bath sonicator (or placing the tip of the sonicator in the test tube) and sonicating for 5-10 minutes above the Tc of the lipid. The lipid suspension should begin to clarify to yield a slightly hazy transparent solution. The haze is due to light scattering induced by residual large particles remaining in the suspension. These particles can be removed by centrifugation toyield a clear suspension of SUV. Mean size and distribution is influenced by composition and concentration, temperature, sonication time and power, volume, and sonicator tuning. Since it is nearly impossible to reproduce the conditions of sonication, size variation between batches producedat different times is not uncommon. Also, due to the high degree of curvature of these membranes, SUV are inherently unstable and will spontaneously fuseto form larger vesicles when stored below their phase transition temperature.
French Pressure Cell Method
The method involves the extrusion of MLV at 20,000 psi at 4°C through a small orifice. The method has several advantages over sonication method. The method is simple, rapid, reproducible and involves gentle handling of unstable materials (Hamilton andGuo, 1984). The resulting liposomes are somewhat larger than sonicated SUVs. The drawbacks of themethod are that the temperature is difficult to achieve and the working volumes are relatively small (about50 mL maximum).
2. SOLVENT DISPERSION METHODS
a. Ether Injection Method
A solution of lipids dissolved in diethyl ether or ether/methanol mixture is slowly injected to an aqueous solution of the material to be encapsulated at 55-65°C or under reduced pressure. The subsequent removal of ether under vacuum leads to the formation of liposomes. The main drawbacks of the method are population is heterogeneous (70-190 nm) and the exposure of compounds to be encapsulated toorganic solvents or high temperature.
b. Ethanol Injection Method
A lipid solution of ethanol is rapidly injected to a vast excess of buffer. The MLVs are immediately formed. The drawbacks of the method are that the population is heterogeneous (30-110 nm), liposomes are very dilute, it is difficult to remove all ethanol because it forms azeotrope with water and the possibility of various biologically activemacromolecules to inactivation in the presence of even low amounts of ethanol
c. Reverse Phase Evaporation Method
First water in oil emulsion is formed by brief sonication of a two phase system containing phospholipids in organic solvent (diethylether or isopropylether or mixture of isopropyl ether and chloroform) and aqueous buffer. The organic solvents are removed under reduced pressure, resulting in the formation of a viscous gel. The liposomes are formed when residual solvent is removed by continued rotary evaporation under reduced pressure. With this method high encapsulation efficiency up to 65% can be obtained in a medium of low ionic strength for example 0.01M NaCl. The method has been used toencapsulate small and large macromolecules. The main disadvantage of the method is the exposure of the materials to be encapsulated to organic solvents and to brief periods of sonication.
3. DETERGENT REMOVAL METHOD:
The detergents at their critical micelles concentrations have been used to solubilize lipids. As the detergent is removed the micelles become progressively richer in phospholipid and finally combine to form LUVs. The detergents can be removed by dialysis. The advantages of detergent dialysis method are excellent reproducibility and production of liposome populations which are homogenous in size. The main drawback of the method is the retention of traces of detergent(s) within the liposomes. A commercial device called LIPOPREP (Diachema AG, Switzerland) which is a version of dialysis system is available for the removal of detergents. Other techniques have been used for the removal of detergents: (a) by using Gel Chromatography involving a column of Sephadex G-259
(b) by adsorption or binding of Triton X-100 (a detergent) to Bio-Beads SM-210 (c) by binding of octyl glucoside (a detergent) to Amberlite XAD-2 beads.
Industrial production of liposomes :
Of the several preparation methods described in theliterature, only a few have potential for large scale manufacturing of liposomes. The main issues faced by formulator and production supervisor are presence of organic solvent residues, physical and chemical stability, pyrogen control, sterility, size and size distribution and batch to batch reproducibility. Liposomes for parenteral use should be sterile and pyrogen free. For animal experiments, adequate sterility can be achieved by the passage of liposomes through 400 nm pore size Millipore filters. For human use, precautions for sterility must be taken during the entire preparation process: that is, (1) the raw materials must be sterile and pyrogen free, (2)preparation in sterile system: working areas equipped with laminar flow and (3) use of sterile containers. Some issues related to phospholipids need attention.12,13 The liposomes based on crude egg yolk phospholipids are not very stable. The cost of purified lipids is very high.The liposomes prepared from polymerizable phospholipids are exposed to UV light. The polymerization process takes place in the bilayer(s). Such liposome preparations usually have better storage stability. It should be noted that such materials usually are phospholipid analogues and their metabolic fates have yet to be established.
Detergent Dialysis
A pilot plant under the trade name of LIPOPREPR II-CIS is available from Diachema, AG, Switzerland.The production capacity at higher lipid concentration (80 mg/ml) is 30 ml liposomes/minute. But when lipid concentration is 10-20 mg/ml then up to many liters of liposomes can be produced. In USA, LIPOPREPR is marketed by Dianorm-Geraete.
Microfluidization
A method based on microfuidization/ microemulsification/homogenization was developed for the preparation of liposomes. MICROFLUIDIZERR is available from Microfudics Corporation, Massachusetts, USA. A plot plant based on this technology can product about 20 gallon/minute of liposomes in 50-200 nm size range. The encapsulation efficiency of up to 75% could be obtained. 14Aqueous dispersions of liposomes often have tendency to aggregate or fuse and may he susceptible to hydrolysis and or oxidation. Two solutions have been proposed:
Proliposomes
In proliposomes, lipid and drug are coated onto a soluble carrier to form free-flowing granular material which on hydration forms an isotonic liposomal suspension. The proliposome approach may provide an opportunity for cost-effective large scale manufacturing of liposomes containing particularly lipophilic drugs.
Lyophilization
Freeze-drying (lyophilization) involves the removalof water from products in the frozen state at extremely low pressures. The process is generally used to dry products that are thermolabile and would be destroyed by heat-drying. The technique has a great potential as a method to solve long term stability problems with respect to liposomal stability. It is exposed that leakage of entrapped materials may take place during the process of freeze- drying andon reconstitution. Recently, it was shown that liposomes when freeze-dried in the presence of adequate amounts of trehalose (a carbohydrate commonly found at high concentrations in organismretained as much as 100% of their original contents. It shows that trehalose is an excellent cryoprotectant (freeze-protectant) for liposomes. Freeze-driers range in size from small laboratory models to large industrial units are available from Pharmaceutical quipment Suppliers. Conventional liposomes are typically composed of only phospholipids (neutral and/or negatively charged) and/or cholesterol. These are characterized by a relatively short blood circulation time. To overcome this problem, longcirculating liposomes (also called stealth or sterically stabilized liposomes) have been developed. These stealth liposomes carry a polymer coating to obtainprolonged circulation times. Targeted liposomes (immunoliposomes) may be either conventionally or sterically stabilized and have specific antibodies or antibody fragments on their surface to enhance target site binding. Cationic liposomes are still under development for improving the delivery of genetic material. A number of products based on liposomes have already been approved for marketing (see Tableno.1), and more are awaiting approval. Companies such as ADD Drug Delivery Techologies AG (Switzerland), DepoTech Corporation (USA), Nexstar Pharmaceuticals (USA), Novavax Inc. (USA), The Liposome Com pany Inc. (USA) and Sequus Pharmaceuticals Inc. (USA) are actively involved in the development of liposomal-based drug delivery systems.
EVALUATION STUDIES:
1. VISUAL APPEARANCE:
Liposome suspension can range from translucent to milky, depending on the composition and particle size. If the turbidity has a bluish shade this means that particles in the sample are homogeneous; a flat, gray color indicates that presence of a nonliposomal dispersion and is most likely a disperse inverse hexagonal phase or dispersed microcrystallites. An optical microscope (phase contrast) can detect liposome> 0.3 µm and contamination with larger particles.
2. DETERMINATION OF ENCAPSULATION EFFICIENCY %:
The EE is the actual weight of the drug entrapped in the liposome compared to the initial amount of drug weighed out. The determination of EE % of liposomes using Remote loading method was carried out as described below.
The percentage of drug entrapped was determined after lysis of the prepared liposomes with 1% Triton and sonication for 10 min. The concentration of liposomes in 1% triton was determined spectrophotometrically at 225 nm using a UV–Vis spectrophotometer, (model UV-1601 PC, Schimadzu, Kyoto, Japan). The entrapment efficiency expressed as entrapment percentage was calculated through the following relationship:
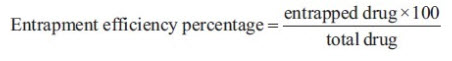
3. DETERMINATION OF LAMILLARITY
The lamellarity of liposomes is measured by electron microscopy or by spectroscopic techniques. Most frequently the nuclear magnetic resonance spectrum of liposome is recorded with and without the addition of a paramagnetic agent that shifts or bleaches thesignal of the observed nuclei on the outer surface of liposome. Encapsulation efficiency is measured by encapsulating a hydrophilic marker
4. LIPOSOME STABILITY
Liposome stability is a complex issue, and consists of physical, chemical, and biological stability. In the pharmaceutical industry and in drug delivery, shelflife stability is also important. Physical stability indicates mostly the constancy of the size and the ratio of lipid to active agent. The cationic liposomes can be stable at 4°C for a long period of time, if properly sterilized.
5. PLASMA STABILITY
Although liposomes resemble biomembranes, they still are foreign objects for the host. Therefore, liposomes are recognized by the mononuclear phagocytic system(MPS) after interaction with plasma proteins. As a result, liposomes are cleared from the blood stream. These stability problems solve by using synthetic phospholipids, gangliosides, polymerization, coating liposomes with chitin derivatives, freeze drying, microencapsulation and particle coated with amphipathic polyethylene glycol.
6. PARTICLE SIZE AND ZETA POTENTIAL:
Zeta potential:
The zeta potential is the electric potential in the interfacial double layer (DL) at the location of the slipping plane versus a point in the bulk fluid away from the interface. In other words, zeta potential is the potential difference between the dispersion medium and the stationary layer of fluid attached to the dispersed particle. For molecules and particles that are small enough, a high zeta potential will confer stability, i.e., the solution or dispersion will resist aggregation. When the potential is low, attraction exceeds repulsion and the dispersion will break and flocculate. So, colloids with high zeta potential (negative or positive) are electrically stabilized while colloids with low zeta potentials tend to coagulate or flocculate .
Particle Size : Determination of Liposomal Size Distribution Size distribution is normally measured by dynamic light scattering. This method is reliable for liposomes with relatively homogeneous size distribution. A simple but powerful method is gel exclusion chromatography, in which a truly hydrodynamic radius can be detected. Sephacryl-S100 can separate liposome in size range of 30-300nm. Sepharose -4B and -2B columns can separate SUV from micelles.
7. IN VITRO DRUG RELEASE STUDY:
The drug-loaded liposomes was placed in the Phosphate buffered saline (PBS) to study the diffusion of the drug from the liposomes. The % of drug-free liposomes can be estimated because of this technique. If the rate of diffusion of the drug is slower in the medium , then it is estimated or confirmed that the formulation is having the highest % encapsulation efficiency. Phosphate buffered saline of pH7.4 is used as a medium of diffusion. It is prepared according to IP monograph.
UV–Vis Spectrophotometry at 225 nm
Drug release from liposomes was studied using a dialysis method. 16mL of the drug-loaded liposomal formulations were placed in a dialysis bag (Spectra/Por dialysis membrane, 12,000–14,000 molecular weight), which was immersed in 50.0 mL phosphate buffer (pH 7.4). The temperature and stirring rate were 37°C and 50 rpm, respectively. Aliquots of 1 ml of the release medium were withdrawn for analysis at 1 hour intervals and replaced with fresh medium. Release runs were continued for a period of 72hours. The absorbance of the collected samples was measured at 225 nm.
8. MOISTURE CONTENT:
In addition to the determination of the pH, weighing and acid-base titration, the determination of the water content is one of the most frequently used methods in laboratories around the world. In contrast to drying, this is a specific method. If no side reactions occur only water will be determined. The method is rapid (normally a few minutes), can be validated and therefore fully documented. The name Karl Fischer (1901–1958), KF for short, is as well known in most laboratories as that of Robert Wilhelm Bunsen (1811 – 1899) and Justus von Liebig (1803 – 1873).With the KF titration both free and bound water can be determined, e.g. surface water on crystals or the water contained inside them. The method works over a wide concentration range from ppm up to 100% and supplies reproducible and correct results.
The KF Reaction:
1. CH3OH + SO2 + RN è[RNH]SO3CH3
2. H2O + I2 + [RNH]SO3CH3 + 2RN è [RNH]SO4CH3 + 2[RNH]I
Basic ingredients of KF reagents : Iodine , Sulphur dioxide , Buffer - Imidazole, Solvent –Methanol
Volumetric KF titration step by step:
1. Fill titration vessel with solvent
2. Pretitration with KF reagent
3. Add the sample
4. Titrate with KF reagent
Volumetric KF reagents :
* One component reagents :
– Titrant contains iodine, sulphur dioxide, buffer and methanol/ethanol
– Working medium contains only the methanol/ethanol
– Disadvantage : the titre decreases 5% per year in the closed bottle!
* Two component reagents :
– Titrant contains iodine
– Solvent contains buffer and sulphur dioxide
– Advantages : pH optimum in the solvent / fast reaction / titre is very stable
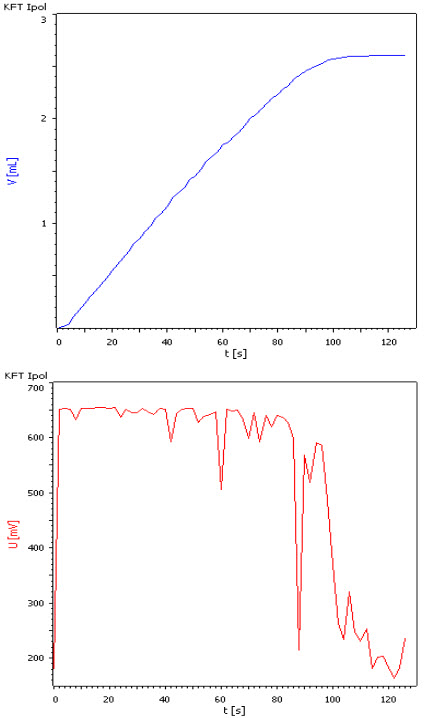
Endpoint indication: During titration: Excess H2O
→ High voltage between Pt wires
End point is not sufficient as criteria (titration would stop at signal at 88 sec)
-> Additionally drift as EP criteria
Figure 3. Endpoint indication
TYPES OF TARGETING :
a. Passive targeting:
Asa mean of passive targeting such usually administered liposomes have been shown to be rapidly cleared off from the blood stream and taken up by the RES in the liver and spleen.Thus capacity of macrophages can be exploited when liposomes are to be targeted by the macrophages.This has been demonstrated by successful delivery of liposomes antimicrobial agents to macrophages.
Liposomes now have been used for targetting of antigens to macrophages as a first step in the index of immunity.for eg. In rats the i.v administration of liposomal antigen elicited spleen phagocyte mediated antibody response whereas the non liposome associated antigen failed to elicit antibody response.
b. Active targeting:
A prerequisite for targeting is the targeting agents are positioned on the liposomal surface such that the interaction with the target i.e.,the receptor is tabulated as aplug and a socket device.The liposome physically prepared such that the lipophillic part of the connector is anchored into the membrane during the formation of the membrane.the hydrophilic part on the surface of the liposome to which the targeting agent should be held in a stearically correct position to bond to the receptor on the cell surface.the active targeting can be brought about by using.
1. Immune liposomes: these are the conventional and stealth liposomes with attached antibodies or other recognition sequence. [eg. Carbohydrate determinants like glycoprotein]the antibody bound , direct the liposome to specific antigenic receptors located on a particular cell.glycoprotein or glycolipid cell surface component that play a role in cell-cell recognition and adhesion.
2. Magnetic liposomes: contain magnetic iron oxide.these liposomes can be directed by an external vibrating magnetic field in their delivery sites.
3. Temperature or heat sensitive liposomes: made in such a way that their transition temperature is just above their body temperature.after reaching the site externally heated the site to release the drug.
APPLICATIONS:
Liposomes can also be decorated with opsonins and ligands to activate endocytosis in other cell types.
- The use of liposomes for transformation or transfection of DNA into a host cell is known as lipofection.
- In addition to gene and drug delivery applications, liposomes can be used as carriers for the delivery of dyes to textiles, pesticides toplants, enzymes and nutritional supplements to foods and cosmetics to the skin.
The use of liposomes in nano cosmetology also has many benefits, including improved penetration and diffusion of active ingredients, selective transport of active ingredients; longer release time, greater stability of active ingredients, reduction of unwanted side effects and high biocompatibility.
Another interesting property of liposomes is their natural ability to target cancer. The endothelial wall of all healthy human blood vessels is encapsulated by endothelial cells that are bound together by tight junctions. These tight junctions stop any large particle in the blood from leaking out of the vessel. Tumour vessels do not contain the same level of seal between cells and are diagnostically leaky. This ability is known as the Enhanced Permeability and Retention effect. Liposomes of certain sizes, typically less than 400nm, can rapidly enter tumoursites from the blood, but are kept in the bloodstream by the endothelial wall in healthy tissue vasculature. Both hydrophilic and hydrophobic drugs can be encapsulated in liposomes. Liposomes are also relatively non-toxic and biodegradable. They therefore have a wide range of biomedical applications.
1. Liposomes in anticancer therapy:
Numerous different liposome formulations of numerousanticancer agents were shown to be less toxic than thefree drug [56-59]. Anthracyclines are drugs which stopthe growth of dividing cells by intercalating into theDNA and, thus, kill mainly rapidly dividing cells. Thesecells are not only in tumors but are also in hair, gastrointestinal mucosa, and blood cells; therefore, this class ofdrug is very toxic. The most used and studied isAdriamycin (commercial name for doxorubicin HCl;Ben Venue Laboratories, Bedford, Ohio). In addition tothe above-mentioned acute toxicities, its dosage islimited by its increasing cardio toxicity. Numerous diverse formulations were tried. In most cases, the toxicitywas reduced to about 50%. These include both acuteand chronic toxicities because liposome encapsulationreduces the delivery of the drug molecules towards thosetissues. For the same reason, the efficiency was in manycases compromised due to the reduced bioavailability ofthe drug, especially if the tumor was not phagocytic orlocated in the organs of mononuclear phagocytic system. In some cases, such as systemic lymphoma, the effect ofliposome encapsulation showed enhanced efficacy due tothe continued release effect, i.e., longer presence oftherapeutic concentrations in the circulation [60-62],while in several other cases, the sequestration of thdrug into tissues of mononuclear phagocytic system actually reduced its efficacy.
Applications in man showed, in general, reduced toxicity and better tolerability of administration with not tooencouraging efficacy. Several different formulations are indifferent phases of clinical studies and show mixed results.
2. Applications of liposomes in medicine and pharmacology:
Applications of liposomes in medicine and pharmacology can be divided into diagnostic and therapeuticapplications of liposomes containing various markers ordrugs, and their use as a tool, a model, or reagent in thebasic studies of cell interactions, recognition processes,and mode of action of certain substances .Unfortunately, many drugs have a very narrow therapeutic window, meaning that the therapeutic concentration is not much lower than the toxic one. In severalcases, the toxicity can be reduced or the efficacy can beenhanced by the use of a suitable drug carrier whichalters the temporal and spatial delivery of the drug, i.e.,its biodistribution and pharmacokinetics. It is clear frommany pre-clinical and clinical studies that drugs, forinstance antitumor drugs, parceled in liposome demonstration reduced toxicities, while retentive enhancedefficacy.
Advances in liposome design are leading to newapplications for the delivery of new biotechnologyproducts, for example antisense oligonucleotides, clonedgenes, and recombinant proteins. A vast literature definethe viability of formulating wide range of conservativedrugs in liposomes, frequently resultant in improvedtherapeutic activity and/or reduced toxicity comparedwith the free drug. As a whole, changed pharmacokinetics for liposomal drugs can lead to improved drug bioavailability to particular target cells that live in thecirculation, or more prominently, to extravascular disease sites, for example, tumors. Recent improvementsinclude liposomal formulations of all-trans-retinoic acid and daunorubicin , which has received Food and Drug Administration consent as a first-linetreatment of AIDS-related advanced Kaposi's sarcoma.Distinguished examples are vincristine, doxorubicin, andamphotericin B .
3. Liposomes in parasitic diseases and infections:
From the time when conventional liposomes aredigested by phagocytic cells in the body after intravenous management, they are ideal vehicles for the targetingdrug molecules into these macrophages. The best knowninstances of this ‘Trojan horse-like’ mechanism are several parasitic diseases which normally exist in the cell ofMPS. They comprise leishmaniasis and several fungal infections. Leishmaniasis is a parasitic infection of macrophages which affects over 100 million people in tropical regions and is often deadly. The effectual dose of drugs, mostly different antimonials, is not much lower than the toxic one. Liposomes accumulate in the very same cell population which is infected, and so an ideal drug delivery vehicle was proposed [52]. Certainly, the therapeutic index was increased in rodents as much as several hundred times upon administration of the drug in various liposomes. Unexpectedly, and unfortunately, there was not much interest to scale up the formulations and clinically approve them after several very encouraging studies dating back to 1978. Only now, there are several continuing studies with various anti-parasitic liposome formulations in humans. These formulations use mostly ionosphere amphotericin B and are transplanted from very successful and prolific area of liposome formulations in antifungal therapy.
4. Protection against enzyme degradation of drugs :
Liposomes are used to protect the entrapped drug against enzymatic degradation whilst in circulation. The basis is that the lipids used in their formulation are not susceptible to enzymatic degradation; theentrapped drug is thus protected while the lipid vesicles are in circulation in the extracellular fluid.
5. Drug targeting :
The approach for drug targeting via liposomes involves the use of ligands (e.g., antibodies, sugar residues, apoproteins or hormones), which are tagged on the lipid vesicles. The ligand recognises specific receptor sites and, thus, causes the lipid vesicles to concentrate at such target sites. By this approach the otherwise preferential distribution of liposomes into the reticuloendeothelial system RES (liver, spleen and bone marrow) is averted or minimized.
6. Topical drug delivery :
The application of liposomes on the skin surface has been proven to be effective in drug delivery into the skin. Liposomes increase the permeability of skin for various entrapped drugs and at the same time diminish the side effect of these drugs because lower doses are now required.
7. Treatment of human immunodeficiency virus (HIV) infections :
Several antiretroviral nucleotide analogues have been developed for the treatment of patients suffering from the acquired immunodeficiency syndromes (AIDS). These include antisense oligonucleotide, which is a new antiviral agent that has shown potential therapeutic application against HIV-1.
8. Enhanced antimicrobial efficacy/ safety :
Antimicrobial agents have been encapsulated in liposomes for two reasons. First, they protect the entrapped drug against enzymatic degradation. For instance, the penicillins and cephalosporin are sensitive to the degradative action of β-lactamase,which is produced by certain microorganisms. Secondly, the lipid nature of the vesicles promotes enhanced cellular uptake of the antibiotics into the microorganisms, thus reducing the effective dose and the incidence of toxicity as exemplified by the liposomal formulation of amphotericin B.
NEW DRUGS:
As of 2008, 11 drugs with liposomal delivery systems have been approved and six additional liposomal drugs were in clinical trials.
List of clinically approved liposomal drugs
|
Name |
Trade name |
Company |
Indication |
|
Liposomal amphotericin B |
Abelcet |
Enzon |
Fungal infections |
|
Liposomal amphotericin B |
Ambisome |
Gilead Sciences |
Fungal and protozoal infections |
|
Liposomal cytarabine |
Depocyt |
Pacira (formerly SkyePharma) |
Malignant lymphomatous meningitis |
|
Liposomal daunorubicin |
DaunoXome |
Gilead Sciences |
HIV-related Kaposi’s sarcoma |
|
Liposomal doxorubicin |
Myocet |
Zeneus |
Combination therapy with cyclophosphamide in metastatic breast cancer |
|
Liposomal IRIV vaccine |
Epaxal |
Berna Biotech |
Hepatitis A |
|
Liposomal IRIV vaccine |
Inflexal V |
Berna Biotech |
Influenza |
|
Liposomal morphine |
DepoDur |
SkyePharma, Endo |
Postsurgical analgesia |
|
Liposomal verteporfin |
Visudyne |
QLT, Novartis |
Age-related macular degeneration, pathologic myopia, ocular histoplasmosis |
|
Liposome-PEG doxorubicin |
Doxil/Caelyx |
Ortho Biotech, Schering-Plough |
HIV-related Kaposi’s sarcoma, metastatic breast cancer, metastatic ovarian cancer |
|
Micellular estradiol |
Estrasorb |
Novavax |
Menopausal therapy |
BENEFITS OF DRUG LOAD IN LIPOSOMES:
|
Benefits of drug load in liposome |
Examples |
|
Amphotericin B, porphyrins, minoxidil, some peptides, and anthracyclines, respectively; hydrophilic drugs, such as anticancer agent doxorubicin or acyclovir |
|
Antimonials, amphotericin B, porphyrins, vaccines, immunomodulators |
|
Doxorubicin, cytosine arabinoside, cortisones, biological proteins or peptides such as vasopressin |
|
Doxorubicin andamphotericin B |
|
Anti-inflammatory drugs, anti-cancer, anti-infection |
|
Antibiotics, chelators, plasmids, and genes |
|
Corticosteroids, anesthetics, and insulin |
FUTURE PROSPECTUS :
Further advances in liposome research have been able to allow liposomes to avoid detection by the body's immune system, specifically, the cells of reticuloendothelial system (RES). These liposomes are known as "stealth liposomes", and are constructed with PEG (Polyethylene Glycol) studding the outside of the membrane. The PEG coating, which is inert in the body, allows for longer circulatory life for the drug delivery mechanism. However, research currently seeks to investigate at what amount of PEG coating the PEG actually hinders binding of the liposome to the delivery site. In addition to a PEG coating, most stealth liposomes also have some sort of biological species attached as a ligand to the liposome in order to enable binding via a specific expression on the targeted drug delivery site. These targeting ligands could be monoclonal antibodies (making an immunoliposome), vitamins, or specific antigens. Targeted liposomes can target nearly any cell type in the body and deliver drugs that would naturally be systemically delivered. Naturally toxic drugs can be much less toxic if delivered only to diseased tissues. Polymersomes, morphologically related to liposomes, can also be used this way. In case of tumor development, certain anticancer drugs such as doxorubicin (Doxil) and daunorubicin are provided through liposomes. Liposomal cisplatin has received orphan drug designation for pancreatic cancer from EMEA.
ATP liposomes.: There is interest in liposomal forms of bioenergic’ substrates, such as ATP, and some encouraging results with ATP-loaded liposomes in various invitro and in vivo models have been reported. ATP liposomes were shown to protect human endothelial cells from energy failure in a cell culture model of sepsis191. In a brain ischaemia model, the use of the liposomal ATP increased the number of ischaemic episodes tolerated before brain electrical silence and death192.
Liposomal haemoglobin.: Active research continues in the area of liposomal haemoglobin (haemosomes) as a blood substitute. To make long-circulating haemosomes, technology for PEG post-insertion was developed, in which the resulting liposomes do not lose any haemoglobin and circulate longer in rabbits183. PEGylated liposomal haemoglobin was found to be stable at storage for 1 year even at room temperature184 and to circulate longer in rabbits when labelled with 99mTc (half-clearance time of48 h)
Liposomes modified with cell-penetrating peptides : .A new approach to drug delivery has recently emerged, which is based on the use of certain viral proteins that have the ability to penetrate into cells (the so-called ‘protein transduction’phenomenon). The transactivating transcriptional activator (TAT) protein from HIV-1 enters various cells when added to the surrounding media.
Magnetic liposomes : An interesting approach for targeted drug delivery under the action of magnetic field is the use of liposomes loaded with a drug and a ferromagnetic material. In one example, magnetic liposomes containing doxorubicin were intravenously administered to osteosarcoma-bearing hamsters.When the tumour-implanted limb was placed between two poles of a 0.4 Tesla magnet, the application of the field for 60 minutes resulted in a fourfold increase in drug concentration in the tumour175. In the same osteosarcoma model in which the magnet was implanted into the tumour, magnetic liposomes loaded with adriamycin demonstrated better accumulation in tumour vasculature and resulted in enhanced tumour-growth inhibition176. Intravenous injection in rats of liposomes loaded with 99mTc-albumin and magnetite resulted in a 25-fold increase in accumulated radioactivity in the right kidney, near which a SmCo magnet was implanted, compared with the control left kidney. This might become a promising way of drug targeting by liposomes.
Cytoskeleton-specific immunoliposomes : Specific anticardiac myosin monoclonal antibodies have an excellent capacity to recognize and bind hypoxic cells with damaged plasma membranes when intracellular myosin is exposed into extracellular space. This property of the antimyosin antibody has been successfully used for the delivery of antibody-bearing liposomes in the field of experimental myocardial infarction. In addition, immunoliposomes specifically targeting ischaemicallydamaged cardiomyocytes (cytoskeleton-specific immunoliposomes) seal membrane damage and decrease the level of cell death both in vitro and in the isolated rat heart model. A similar approach was used for decreasing haemorrhage after focal embolic stroke by antiactin-targeted liposomes in rats. Cytoskeleton-specific immunoliposomes can fuse with damaged cells, and so they were used as carriers for successful gene delivery into hypoxic cells.
CONCLUSION :
Liposomes have been used in a broad range of pharmaceutical applications. Liposomes are showing particular promise as intracellular delivery systems for anti-sense molecules, ribosomes, proteins/peptides, and DNA. Liposomes with enhanced drug delivery to disease locations, by ability of long circulation residence times, are now achieving clinical acceptance. Also, liposomes promote targeting of particular diseased cells within the disease site. Finally, liposomal drugs exhibit reduced toxicities and retain enhanced efficacy compared with free complements. Only time will tell which of the above applications and speculations will prove to be successful. However, based on the pharmaceutical applications and available products, we can say that liposomes have definitely established their position in modern delivery systems.
REFERENCES :
1. Kimball's Biology Pages, "Cell Membranes." Stryer S. Biochemistry, 1981, 213.
2. Vyas S.P. & Khar R.K. “Targeted & controlled drug delivery : Novel carrier system”. CBS publishers and distributors, 2007.
3. Gomez-Hens, A., Fernandez-Romero, J.M. “Analytical methods for the control of liposomal delivery systems”, Trends Anal Chem, 2006,25,167–178.
4. Mozafari, M.R., Johnson, C., Hatziantoniou, S. & Demetzos, C. “ Nanoliposomes and their applications in food nanotechnology”, Journal of Liposome Research, 2008, 18, 309-327.
5. Danilo D. L. “Liposomes in Gene Delivery”. CRC press, 1997.
6. Riaz M.; “review : liposomes preparation methods,” Pak. J. Pharm. Sci.; 1996, 19, 65-77.
7. Deamer D. and Bangham A.D. Biochim. Biophys. Acta.; 1976; 443: 629.
8. Batzri S. and Korn E.D. Biochim. Biophy. Acta.; 1973; 298: 1015.
9. Frank Szoka, Jr. & Demetrios Papahadjopoulos, “Comparative properties and methods of preparation of lipid vesicles (Liposomes),” Ann. Rev. Biophys. Bioeng., 1980, 9, 467-508.
10. Kagawa Y. and Racker E. (1974). J. Biol. Chem. 246: 5477.
11. Enoch H.G. and Strittmatter P. (1979). Proc. Natl. Acad. Sci. USA. 76: 145.
12. Gerritson W.J., Verkley A.J., Zwaal R.F.A. and van Deenan L.L.M. (1978). Eur. J. Biochem. 85: 255.
13. Philippot J.R., Mutafschicv S. and Liautard J.P. (1985). Biochim. Biophys. Acta. 821: 79.
14. Gregoriadis G. Liposome Technology. CRC Press, Boca Raton, Volumes I, II and III, 1984.
15. Gregoriadis G. Liposome Technology 2nd Edition. CRC Press, Boca Raton, Volumes I, II and III, 1992.
16. Maierhofer G. (June 1985). Am. Clinical Products. 33.
17. Mayhew E., Nikolopoulos G.T., King J.J. and Siciliano A.A. (1985). Pharm. Manufacturing.2: 18.
18. Payne N.I. Browning I. and Hynes C.A. (1986). J. Pharm. Sci. 75: 330.
19. Crow J.H., Spargo B.J. and Crow L.M. (1987). Proc. Natl. Acad. Sci. USA. 84: 1537.
20. Rajan K. Verma and Sanjay Garg, “Current Status of Drug Delivery Technologies and Future Directions,” Pharmaceutical Technology On-Line, 25 (2), 1–14 (2001).
21. Barani, H. & Montazer, M. “A Review on Applications of Liposomes in Textile Processing. Journal of Liposome Research,” 2008, 18, 249-262.
22. Kamble R., Pokharkar V. B., Badde S and Kumar A (2010). Development and characterization of liposomaldrug delivery system for nimesulide. Int. J. Pharm. Pharm. Sci., 2; 4: 87-89.
23. Bangham A. D (1983). Liposomes. Marcel Dekker, New York, Ed. Ist : 1-26. 3. Jain N. K (2007). Controlled and novel drug delivery. CBS publishers and distributors, New Delhi: 304.
24. Patil S. G., Gattani S. G., Gaud R. S., Surana S. J., Dewani S. P. and Mahajan H. S (2005). The Pharma Review, 18(3):53-58.






