

{ DOWNLOAD AS PDF }
ABOUT AUTHORS
Kambham Venkateswarlu*, Jami Komala Preethi
1Department of Pharmaceutics, JNTUA-Oil Technological and Pharmaceutical Research Institute, Jawaharlal Nehru Technological University Anantapur, Ananthapuramu, Andhra Pradesh, India.
k.v.reddy9441701016@gmail.com
ABSTRACT
The present investigation was focussed on formulation and in vitro evaluation of mouth dissolving tablets (MDTs) of Pioglitazone Hydrochloride (PGTZN) thereby enhancing the dissolution rate. MDTs were prepared by wet granulation and direct compression methods using Croscarmellose sodium, Crospovidone and Sodium starch glycolate as superdisintegrants. Powder blends were evaluated for flowability and all the powder blends showed acceptable flowability. The prepared tablets were evaluated for post compression parameters like hardness, friability, wetting time and showed acceptable results. Formulations F8 and F15 showed disintegration time of 23 and 22 sec respectively. Dissolution was performed in pH 1.2 HCl buffer and formulations F15 showed maximum drug release within 30 min. drug release from F15 was more than that of the marketed drug. Hence, it could be concluded that formulation F15 showed good drug release than marketed drug and there is a lot of scope for future in vivo studies.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-2453
|
PharmaTutor (ISSN: 2347 - 7881) Volume 4, Issue 12 Received On: 16/08/2016; Accepted On: 22/08/2016; Published On: 01/12/2016 How to cite this article: K Venkateswarlu, Jami KP; Formulation development and in vitro evaluation of mouth dissolving tablets of Pioglitazone Hydrochloride; PharmaTutor; 2016; 4(12); 37-42 |
INTRODUCTION
Drug Delivery Systems (DDS) area is the strategic tool for expanding markets/indications, extending product life cycles and generating opportunities. DDS make a significant contribution to global pharmaceutical sales through market segmentation, and are moving rapidly. Despite of tremendous advancements in drug delivery, the oral route remains the perfect route for the administration of therapeutic agents because of low cost of therapy, ease of administration, accurate dosage, self‐medication, pain avoidance, versatility, leading to high levels of patient compliance.[1] Improved patient compliance has achieved enormous demand. Consequently demand for their technologies is also increasing manifolds. To develop a chemical entity, a lot of money, hard work and time are required. Hence, focus is rather being laid on the development of new drug delivery systems for already existing drugs, with enhanced efficacy and bioavailability, thus reducing the dose and dosing frequency to minimize the side effects.[2] It is always the aim of a scientist or a dosage form designer to enhance the safety of a drug molecule while maintaining its therapeutic efficacy. Recent advances in Novel Drug Delivery Systems (NDDS) aim for the same by formulating a dosage form, convenient to be administered so as to achieve better patient compliance.[3] Pharmaceutical technologists have put in their best-efforts to develop a Fast Dissolving Drug Delivery System i.e. Mouth Dissolving Tablet (It is a tablet that disintegrates and dissolves rapidly in the saliva, within a few seconds without the need of drinking water or chewing). A mouth dissolving tablet usually dissolves in the oral cavity within 15 sec to 3 min. Most of the MDTs include certain super disintegrants and taste masking agents.[4]
Diabetes Mellitus (DM) is a group of syndromes and chronic metabolic disorder characterized by hyperglycemia, altered metabolism of lipids, carbohydrates and proteins because of a lack of or ineffective use of the hormone insulin and associated with reduced life expectancy, significant morbidity due to specific diabetes related micro vascular complications and diminished quality of life. A fasting blood glucose level of 126 mg/dl and 200 mg/dl post prandial (oral Glucose load) is considered as indication of DM.[5] In present work, an investigation was made to use crospovidone and sodium starch glycolate, Croscarmellose sodium as superdisintegrants in the design of mouth dissolving tablets.
MATERIALS AND METHODS
FTIR studies
It was performed to know the compatibility between pure drug and excipients used in the formulation development. The physicochemical compatibility between the PGTZN and excipients was carried out by using Perkin Elmer Fourier Transform Infra Red spectrophotometer, Shelton USA. KBr disc method was employed and the samples were scanned from 4000 cm-1 to 400 cm-1. An obtained IR spectrum of pure drug was compared with the IR spectra of its physical mixtures to know the chemical interactions between drug and excipients used in the formulation.[6]
Preparation of PGTZN MDTs by direct compression method
Tablets containing 15 mg of PGTZN were prepared by direct compression method and the various formulae used in this study are shown in table 1. The drug and diluents were passed through sieve on 40. All the above ingredients were properly mixed together (in a poly-bag). Talc and magnesium stearate were passed through mesh number 80, mixed well and blended with initial mixture in a poly-bag followed by compression of the blend using Rotary tablet punching machine, station 10, Elite scientific and equipment, India.
Preparation of PGTZN MDTs by wet granulation method
Tablets containing 15 mg of PGTZN were prepared by wet granulation method and the various formulae used in the study are shown in the table 1. The drug and diluent were passed through sieve no 12 and all the above ingredients were properly mixed together (in a poly-bag). Talc and magnesium stearate were passed through mesh number 80, mixed well and blended with intial mixture in a poly-bag followed by compression of the blend using Rotary tablet punching machine, station 10, Elite scientific and equipment, India.
Table 1. Preparation of PGTZN MDTs
|
Direct compression |
Wet granulation |
||||||||||||||
|
Ingredients |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
F11 |
F12 |
F13 |
F14 |
F15 |
|
PGTZN |
15 |
15 |
15 |
15 |
15 |
15 |
15 |
15 |
15 |
15 |
15 |
15 |
15 |
15 |
15 |
|
Croscarmellose sodium |
4.5 |
|
|
3 |
3 |
|
4.5 |
4.5 |
- |
4.5 |
- |
3 |
3 |
3 |
4.5 |
|
Crospovidone |
- |
4.5 |
|
3 |
- |
3 |
4.5 |
- |
4.5 |
4.5 |
4.5 |
3 |
- |
- |
- |
|
Sodium starch glycolate |
- |
|
4.5 |
- |
3 |
3 |
- |
4.5 |
4.5 |
- |
4.5 |
- |
3 |
3 |
4.5 |
|
Microcrystalline cellulose |
94 |
94 |
94 |
93 |
93 |
93 |
90 |
90 |
90 |
90 |
90 |
93 |
93 |
93 |
90 |
|
Lactose |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
30 |
|
Talc |
2.5 |
2.5 |
2.5 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
|
Magnesium stearate |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
|
Aspartame |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
|
Total weight |
150 |
150 |
150 |
150 |
150 |
150 |
150 |
150 |
150 |
150 |
150 |
150 |
150 |
150 |
150 |
Evalution of tablets
Determination of precompression characteristics
The precompression parameters like Angle of repose,[7] Carr’s Index, Hausner’s ratio, Dispersibility[8] and Porosity (Ø)[9] were performed to know the powder characteristics.
Determination of post compression characteristics
The post compression parameters like thickness, friability,[10] hardness, weight variation,[11] disintegration time,[12] wetting time[2] and drug content [13] were evaluated for the formulated tablets.
[adsense:468x15:2204050025]
Dissolution studies
The release rate of PGTZN was determined by using USP dissolution apparatus type-II (Paddle) (Electro Lab TDT-06N USP dissolution apparatus, India). The dissolution test was performed using 900 ml of pH 6.8 phosphate buffer solution for 30 min which was maintained at 37 ± 0.2°C and rate of stirring at 50 rpm. At each time point of analysis, 5 ml sample was withdrawn and replaced with freshly prepared dissolution media maintained at same conditions. Sample solution was filtered, drug content of the filtrate was determined spectrophotometrically.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
RESULTS AND DISCUSSION
MDTs of PGTZN were formulated by wet granulation and direct compression methods using Croscarmellose, Crospovidone and Sodium starch glycolate as disintegrating agents, microcrystalline cellulose and lactose as diluents. The PGTZN and sulfonylurea are given in combination for treatment of type-2 Diabetes mellitus for long term therapy. During this therapy, it is observed that there is uncontrolled increase of blood glucose level due to the hepatic metabolism of drugs. Therefore, MDTs of PGTZN were prepared to overcome this unusual problem i.e, to protect the drug from gastric secretion and enhance the absorption.[5]
FTIR studies
The IR spectrum of pure drug and its physical mixture were studied. The characteristic absorption peaks of PGTZN like N-H str, C-H str, C-O Str and C-H ben were obtained at wave numbers 3474.20 cm-1, 3085.05 cm-1, 1149.01 cm-1, 849.66 cm-1 respectively and also those peaks were observed in IR spectra of physical mixtures (Figure 1). Hence, it indicates there were no chemical interactions between the pure drug and its physical mixture.
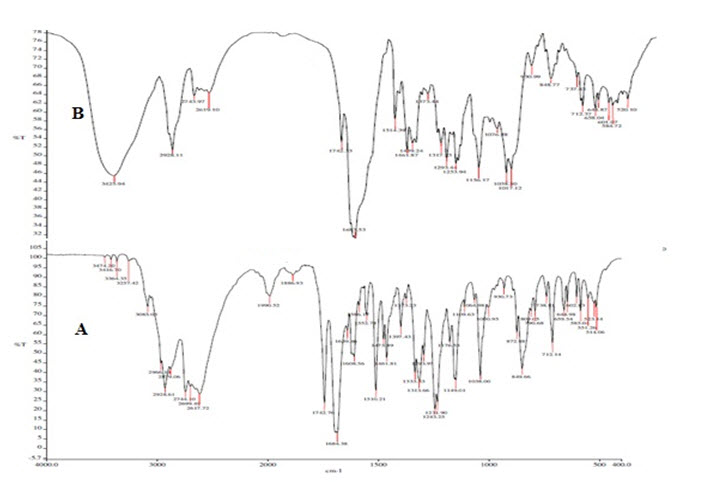
Figure 1. FTIR studies. A) IR spectra of pure drug, B) IR spectra of physical mixture
Precompression studies
The powder blends have the bulk density ranged from 0.290 to 0.456 and tapped density from 0.330 to 0.569. The compressibility index values less than 10, 11-15, 16-20, 21-25, 26-31, 32-37 and greater than 38 indicates excellent, good, fair, possible, poor, very poor and very very poor flow respectively and powder blends of all the formulations were found to be possessing the excellent to fair flowability i.e. 5.1 to 18.18 %. The ideal Hausner’s ratio values of 1.0-1.11, 1.12-1.18, 1.19-1.25, 1.26-1.34, 1.35-1.45, 1.46-1.59 and greater 1.60 indicates excellent, good, fair, possible, poor, very poor and very very poor flow respectively and powder blends of all the formulations were found to be possessing the excellent to fair flowability i.e. 1.05 to 1.22. The ideal angle of repose values of less than 25, 25-30, 30-40 and greater than 40 indicates excellent, good, passable and very poor respectively and the experimental values of all the formulations were ranged from 27.92º to 37.59º indicates good to passable flowability. Powder blend also showed acceptable dispersibility and porosity results (Table 2).
Table 2. Precompression Parameters
|
Direct compression |
Wet granulation |
|||||||||||||||
|
Parameters |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
F11 |
F12 |
F13 |
F14 |
F15 |
Marketed drug |
|
Bulk density (gm/cc) |
0.456 |
0.452 |
0.447 |
0.456 |
0.452 |
0.448 |
0.462 |
0.445 |
0.398 |
0.29 |
0.34 |
0.37 |
0.40 |
0.36 |
0.35 |
0.446 |
|
Tapped density (gm/cc) |
0.543 |
0.540 |
0.552 |
0.553 |
0.567 |
0.539 |
0.547 |
0.569 |
0.548 |
0.33 |
0.36 |
0.39 |
0.43 |
0.44 |
0.37 |
0.36 |
|
Porosity% |
18.3 |
26.7 |
28 |
18.2 |
26.6 |
28 |
18.3 |
25 |
28 |
16.2 |
24.2 |
24.17 |
24 |
25.2 |
18.2 |
28 |
|
Carr’s index |
16.7 |
15.37 |
14.17 |
14.19 |
15.17 |
16.18 |
16.96 |
17.02 |
15.19 |
12.12 |
5.5 |
5.1 |
16.9 |
18.18 |
5.40 |
5.41 |
|
Hausner’s ratio |
1.173 |
1.21 |
1.18 |
1.173 |
1.192 |
1.204 |
1.189 |
1.186 |
1.172 |
1.13 |
1.058 |
1.05 |
1.07 |
1.22 |
1.05 |
1.172 |
|
Dispersibility (%) |
70.3 |
68.3 |
70.3 |
82.5 |
75.4 |
68.3 |
70.3 |
56.8 |
66.3 |
80.5 |
65.3 |
68.2 |
54.2 |
65.2 |
70.2 |
70.2 |
|
Angle of repose (0) |
31.21 |
29.69 |
30.21 |
31.33 |
30.14 |
35.23 |
29.14 |
28.22 |
29.13 |
37.23 |
29.68 |
27.92 |
34.21 |
37.59 |
28.69 |
31.4 |
Post compression parameters like hardness, friability, weight variation, disintegration time and wetting time were studied and reported in table 3. Hardness was ranged from 3.5 to 5 kg/cm2 and supports sufficient mechanical strength to the tablets. Friability was lies between 0.25 to 0.95 % and complied with the pharmacopoeial limits. Tablets posses uniform thickness and passed weight variation test. Drug content showed by all the formulations was complied with the official limits and results were very close to the marketed drug.
Disintegration time
The most important parameter that is need to be optimized during the development of MDTs is disintegrating time of tablets (Table 3). The disintegration test of the tablets was conducted in pH 6.8 phosphate buffer and showed disintegration time from 22 to 32 sec. However, disintegration time of the tablets prepared with of superdisintegrants (1:1) is in the acceptable range with no much deviation due to the combinational effect of super disintegrant mixture. Comparatively, the disintegration time of the prepared tablets are in the order as F15<F8<F11<F9<F10<F13=F5 <F14<F7<F12<F6=F4=F2<F1.
Wetting time
Wetting time of tablets containing the mixture of superdisintegrants (1:1) with various concentrations are in the range of 11-24 sec (Table 3). Usually the crospovidone having the low swelling rate has also showed the fast wetting effect due to the combinational effect of superdisintegrants. This is also due to the synergistic effect of crospovidone along with other superdisintegrants will modify the wetting property of crospovidone and formulations prepared with the mixture of it. It was observed that there was no significant change in the wetting time with increase in the concentration of superdisintegrants mixture and however, the formulations with mixture of superdisintegrants like Croscarmellose sodium and Sodium starch glycolate showed least disintegration time. The wetting of tablets are in the following order F15<F8<F10=F11<F9<F13 <F14<F7<F5<F12<F6<F4<F3<F1<F2.
Table 3. Evaluation of post compression parameters
|
Direct compression |
Wet granulation |
|||||||||||||||
|
Parameters |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
F11 |
F12 |
F13 |
F14 |
F15 |
Marketed drug |
|
Hardness (kg/cm2) |
3.7 |
3.9 |
3.8 |
3.6 |
4.8 |
4.9 |
5.0 |
4.1 |
3.9 |
3.5 |
4.6 |
4.8 |
4.9 |
4.0 |
3.63 |
3.9 |
|
Friability (%) |
0.68 |
0.69 |
0.67 |
0.75 |
0.69 |
0.43 |
0.21 |
0.97 |
0.14 |
0.74 |
0.68 |
0.40 |
0.20 |
0.95 |
0.75 |
0.21 |
|
Weight variation |
Pass |
Pass |
pass |
Pass |
Pass |
Pass |
Pass |
Pass |
Pass |
Pass |
Pass |
Pass |
Pass |
Pass |
Pass |
pass |
|
Thickness (mm) |
4.23 |
4.22 |
4.21 |
4.28 |
4.35 |
4.33 |
4.25 |
4.25 |
4.24 |
4.25 |
4.33 |
4.32 |
4.20 |
4.32 |
4.23 |
4.25 |
|
Disintegration time (sec) |
34 |
32 |
31 |
32 |
28 |
32 |
30 |
23 |
26 |
27 |
24 |
31 |
28 |
29 |
22 |
24 |
|
Wetting time (sec) |
26 |
28 |
25 |
24 |
20 |
23 |
18 |
13 |
15 |
14 |
14 |
22 |
16 |
17 |
11 |
29 |
|
Drug content (%) |
98.3 |
98.5 |
99.6 |
98.5 |
99.6 |
98.9 |
100.2 |
98.0 |
101 |
96.5 |
98.6 |
96.9 |
99.2 |
100.4 |
102.0 |
102.4 |
The rapid increase in dissolution (Table 4) of PGTZN in F15 and F8 may be due to the rapid swelling of Croscarmellose and sodium starch glycolate. The increase in the concentration of Croscarmellose and sodium starch glycolate increased the swelling and disintegration of tablets rapidly into apparently small particles. While tablets formulated with sodium starch glycolate disintegrated by rapid up take of buffer, followed by rapid and enormous swelling into primary particle but more rapidly due to the change in viscous gel layer of sodium starch glycolate by Croscarmellose. The % drug release of all the formulations are in the order: F15>F8>F11>F9 >F10>F13>F14>F7>F5>F12>F6> F4>F3>F1>F2.
Table 4. In-vitro dissolution profiles of PGTZN MDTs
|
Direct compression |
Wet granulation |
|||||||||||||||
|
Time interval (min) |
F1 |
F2 |
F3 |
F4 |
F5 |
F6 |
F7 |
F8 |
F9 |
F10 |
F11 |
F12 |
F13 |
F14 |
F15 |
Marketed drug |
|
5 |
31.42 |
28.32 |
32.22 |
59.26 |
61.73 |
58.57 |
67.71 |
65.41 |
62.85 |
63.71 |
69.42 |
62.87 |
64.21 |
62.57 |
70.85 |
68.31 |
|
10 |
37.52 |
34.22 |
36.43 |
63.834 |
67.82 |
63.98 |
71.33 |
71.32 |
75.04 |
67.86 |
73.07 |
66.42 |
69.02 |
66.42 |
76.52 |
75.43 |
|
15 |
40.52 |
38.33 |
40.22 |
69.44 |
78.86 |
72.64 |
79.48 |
76.31 |
80.91 |
83.46 |
80.94 |
71.10 |
81.46 |
75.41 |
84.131 |
83.18 |
|
20 |
48.32 |
45.09 |
47.43 |
75.74 |
81.59 |
78.6 |
81.65 |
82.47 |
82.79 |
85.93 |
83.11 |
78.67 |
84.21 |
81.85 |
92.93 |
90.15 |
|
25 |
52.44 |
52.11 |
54.33 |
85.43 |
87.49 |
86.25 |
86.40 |
93.88 |
92.73 |
87.55 |
88.16 |
86.28 |
88.40 |
88.33 |
94.004 |
93.33 |
|
30 |
58.24 |
57.42 |
61.49 |
90.72 |
93.00 |
91.03 |
93.33 |
96.22 |
94.38 |
94.35 |
95.53 |
92.21 |
94.34 |
93.71 |
98.53 |
96.77 |
From this study, it can be concluded that wet granulation method showed better disintegration and drug release as compared to direct compression method. The main aim of formulating mouth dissolve tablets was to achieve instantaneous dispersion without the aid of water. By seeing in vitro dissolution time and disintegration, it can be clearly stated that the objective has been achieved. Above all, most of the formulations showed 80% drug release within 30 min, hence decreasing the lag time for absorption. By seeing this, it can be clearly seen that there is more chance for pre gastric absorption, thereby reducing first pass metabolism. Therefore, overall oral bioavailability can be increased. As the target patients are children and elderly, the addition of sweeteners increased the appeal and patient compliance.
REFERENCES
1. Tejvir K., Bhawandeep G., Sandeep K. and Gupta G.D.; Mouth dissolving tablets: a novel app to drug delivery; Int. J. Curr. Pharm. Res.; 2011;3;1-7.
2. Venkateswarlu K. and Chandrasekhar K.B.; Formulation and in-vitro evaluation of lacidipine oral disintegrating tablets: enhancement of solubility and dissolution rate; Int. J. Life. Sci. Pharm. Res.; 2016; 6(2);16-26.
3. Venkateswarlu K., Thirumalesh Naik S.B. and Chandrasekhar K.B.; Formulation and in vitro evaluation of orlistat orodispersible tablets for enhancement of dissolution rate; Int. J. Pharm. Pharm. Sci.; 2016;8(4);236-241.
4. Thirumalesh Naik S.B., Venkateswarlu K. and Chandrasekhar K.B.; Formulation and in-vitro evaluation of orodispersible tablets of olanzapine for the improvement of dissolution rate; J. Chem. Pharm. Res.; 2016;8(1);177-181.
5. Tripathi K.D; Essentials of medicinal pharmacology; Jaypee brother’s medical publishers, Delhi; 2001.
6. Vijayabhaskar K., Venkateswarlu K. and Thirumalesh Naik S.B.; Preparation and in-vitro evaluation of ranitidine mucoadhesive microspheres for prolonged gastric retention; Br. J. Pharm. Res.; 2016;10(2);1-12.
7. Venkateswarlu K., Chandrasekhar K.B. and Ramachandra R.; Development and in-vitro Evaluation of Reconstitutable Suspension of Flucloxacillin; Marmara. Pharm. J.; 20(3); 280-287, 2016.
8. Subramanyam C.V.S; Text book of pharmaceutical formulation; Vallabhprakashan publishers; Delhi; 2007.
9. Rowe R.C., Sheskey P.J. and Owen S.C.; Handbook of Pharmaceutical Excipients, Royal Pharmaceutical Society of Great Britain; Britain; 2006.
10. Thirumalesh Naik S.B., Venkateswarlu K. and Chandrasekhar K.B.; Formulation and in-vitro evaluation of Pregabalin mini tablets for sustained release; Pharm. Lett.; 2016;8(2);277-283.
11. Venkateswarlu K. and Chandrasekhar K.B.; Development and Statistical Optimization of Sustained Release Gastro Retentive Floating Tablets of Cephalexin; Marmara. Pharm. J.; 2016; 22(2);172-183.
12.Thirumalesh Naik S.B, Venkateswarlu K. and Chandrasekhar K.B.; Formulation and evaluation of Oxybutynin chloride extended release matrix tablets; IndoAm. J. Pharm. Res.; 2016;6(01);4179-4184.
13. Venkateswarlu K. and Shanthi A.; Formulation and evaluation of Glipizide matrix tablets; IOSR J. Pharm. Biol. Sci.; 2012;2(5);17-23.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






