

{ DOWNLOAD AS PDF }
ABOUT AUTHORS
Rohit A. Patil1*, Shruti N.Patil2
Department of Regulatory Affairs,
Supreme Pharma Healthcare Pvt. Ltd. Mumbai, MH, India
* rohitpharma3250@gmail.com
ABSTRACT
Good storage of data & record management are critical elements of pharmaceutical quality system. Data integrity refers to maintaining & assuring the accuracy & consistency of data over its entire life –cycle in compliance with its applicable regulatory requirements.
Data integrity is mandatory for the regulated pharmaceutical industry, as processing and disposition decisions regarding product quality, safety, efficacy, purity, and compliance with the applicable regulatory requirements are made based on data that is recorded and reported. Data integrity risk should be assessed, mitigated, communicated & reviewed throughout the data life cycle. Healthcare industries should be designed Record-keeping methodologies and systems, in a way that encourages compliance and assures data quality and reliability.
[adsense:336x280:8701650588]
REFERENCE ID: PHARMATUTOR-ART-2407
|
PharmaTutor (Print-ISSN: 2394 - 6679; e-ISSN: 2347 - 7881) Volume 4, Issue 5 Received On: 26/11/2015; Accepted On: 10/12/2015; Published On: 01/05/2016 How to cite this article: Patil RA, Patil SN; Data Integrity and Worldwide Regulatory Guidance; PharmaTutor; 2016; 4(5); 20-24 |
INTRODUCTION
Data integrity is the assurance that data records are accurate, complete, intact and maintained within their original context, including their relationship to other data records.
This definition applies to data recorded in electronic and paper formats or a hybrid of both. A good way to understand data integrity is through an analogy from the legal world. A data record is similar to a contract. A contract is valid only if all the pages of the document are complete and legible, contain the required, authentic signatures and properly state the terms and conditions. In this sense, integrity denotes validity.[1]
Ensuring data integrity means protecting original data from accidental or intentional modification, falsification or even deletion, which is the key to reliable and trustworthy records that will withstand scrutiny during regulatory inspections.[1]
“The sum total of arrangements to ensure that data, irrespective of the format in which it is generated, is recorded, processed, retained and used to ensure a complete, consistent and accurate record throughout the data lifecycle”[2]
MAIN CRITERIA FOR DATA INTEGRITY:[3]
• The quality of correctness, completeness, wholeness, soundness and compliance with the intention of the creators of the data.
• It is achieved by preventing accidental or deliberate but unauthorized insertion, modification or destruction of data in a database.
• Data integrity is one of the six fundamental components of information security.
US FDA- ALCOA[2]
According to the FDA, data should meet certain fundamental elements of quality. Whether they’re recorded on paper or electronically, source data should be attributable, legible, contemporaneous, original, and accurate (ALCOA) [2]
Table No: 1[2]
|
Accurate |
No errors or editing without documented amendments |
|
Attributable |
Who acquired the data or performed an action and when? |
|
Attributable |
Who acquired the data or performed an action and when? |
|
Complete |
All data is present and available |
|
Consistent
|
All elements of the record, such as the sequence of events, follow on and are dated or time stamped in expected sequence |
|
Contemporaneous |
Documented at the time of the activity |
|
Enduring |
On proven storage media (paper or electronic) |
|
Legible |
Can you read the data? |
|
Original/Reliable |
Written printout or observation or a certified copy thereof |
|
Trustworthy |
The data and the record have not been tampered with |
If these best practices for source documentation aren’t followed, there is no valid evidence that the test article is safe and effective. Data integrity is the assurance that data records are accurate, complete, intact and maintained within their original context, including their relationship to other data records. This definition applies to data recorded in electronic and paper formats or a hybrid of both.
[adsense:468x15:2204050025]
DATA SHOULD BE (ALCOA):
ATTRIBUTABLE
Is it traceable to a person, date, and subject visit?
When documenting data on paper, every written element needs to be traced back to the authorized individual who is responsible for recording it. This requires a signature or initials, the date, and an identifier to a subject visit. Similarly, if something needs to be changed on the record, it needs to be initialed, dated, and should explain the reason for the change.
Audit trails in an electronic system make it very obvious who created a record, when it was created, who made a change, when the change was made, and the reason a change was made. A compliant system will automatically track this information and enable electronic signatures. Data is attributable to a unique user with a secure password and role-based permissions, preventing changes from being made by unauthorized users.
LEGIBLE
Is it clear enough to read?
On paper, everything that’s written must be easy to read and recorded in a permanent medium (not pencil). Handwriting must be clear to reduce the likelihood of transcription errors and allow a study to be accurately re-created.
Electronic source records typically solve the illegible handwriting problem, because data and information are presented in a clean and standardized format.
CONTEMPORANEOUS
Was it recorded as it happened?
Data should be recorded, signed, and dated at the time of trial conduct, rather than risk an individual recalling the wrong information from memory. On paper, data needs to be documented in real-time and dated with the current date (no predating or postdating).
Automatic date and time stamps support this every time data is entered, edited, or modified in an electronic system.
ORIGINAL
Is it the first place data is recorded?
The source is the earliest record – the first place that data is documented. If corrections or revisions need to be made, changes shouldn’t obscure prior entries. Paper source documents should be preserved and kept in their original form.
When the first record is electronic, an audit trail can track any and all subsequent queries and changes.
ACCURATE
Are all the details correct?
It’s critical that the source completely reflects the true observations. This means an honest, accurate, and thorough representation of facts describing the conduct of the study. There will be times when source documents are incomplete, inconsistent, or wrong. If changes need to be made, modifying a paper record always need to be done in a compliant manner.
When the source is electronic, audit trails can provide transparency to prevent data from being altered in a way that is difficult to detect. Additionally, automatic edit checks can immediately alert when missing data points or out-of-range data are entered.
All of the elements of the acronym ALCOA must be applied to both paper and electronic source data, and the records that hold that data. Serving as evidence of the events that took place during a study, source documents need to paint the full picture of what happened. Using ALCOA as a guide to collecting quality data in clinical trials can help justify that a test article is safe and effective.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
ESTABLISHING DATA CRITICALITY & INHERENT INTEGRITY RISK
In addition to an overarching data governance system, which should include relevant policies and staff training in the importance of data integrity, consideration should be given to the organizational (e.g. procedures) and technical (e.g. computer system access) controls applied to different areas of the quality system.
The degree of effort and resource applied to the organizational and technical control of data lifecycle elements should be commensurate with its criticality in terms of impact to product quality attributes.
Data may be generated by[2]
A paper-based record of a manual observation, or
In terms of equipment, a spectrum of simple machines through to complex highly configurable computerized systems.
The inherent risks to data integrity may differ depending upon the degree to which data (or the system generating or using the data) can be configured, and therefore potentially manipulated (see figure 1).
SIMPLE VS COMPLEX – A RISK BASED APPROACH[2, 4]

Figure 1: Diagram to illustrate the spectrum of simple machine (left) to complex computerized system (right) & relevance of printouts as ‘original data’[2,4]
With reference to figure 1, simple systems (such as pH meters and balances) may only require calibration, whereas complex systems require ‘validation for intended purpose’. Validation effort increases from left to right in the diagram above. However, it is common for companies to overlook systems of apparent lower complexity. Within these systems it may be possible to manipulate data or repeat testing to achieve a desired outcome with limited opportunity of detection (e.g. stand-alone systems with a user configurable output such as FT-IR, UV spectrophotometers).
DESIGNING SYSTEMS TO ASSURE DATA QUALITY AND INTEGRITY
Systems should be designed in a way that encourages compliance with the principles of data integrity. Examples include:
- Access to clocks for recording timed events
- Accessibility of batch records at locations where activities take place so that ad hoc data recording and later transcription to official records is not necessary
- Control over blank paper templates for data recording
- User access rights which prevent (or audit trail) data amendments
- Automated data capture or printers attached to equipment such as balances
- Proximity of printers to relevant activities
- Access to sampling points (e.g. for water systems)
- Access to raw data for staff performing data checking activities.
Challenges of Maintaining Laboratory Data Integrity[1]
The first challenge in ensuring the integrity of laboratory data involves utilization of hybrid systems (see Figure 2). If both are used, paper and electronic records need to be synchronized.[1]

Figure 2: Data Handling Alternatives[1]
Computerized systems add a second challenge to laboratory data integrity due to the potential for electronic data manipulation. Such manipulation can include human errors when data are entered by mistake or intentionally falsified, and selection of good or passing results to the exclusion of those that are poor or failing. One example of how to overcome some of the challenges of managing data integrity is in the manual entry of critical data. After manual entry of the critical data is complete, a verification of the data entry can be performed by a second person or can be verified with the use of a validated computerized verification process. The third challenge comes from system interfaces. If interfaces are used to transfer data from instrument to system or between systems, such as between the chromatography data system (CDS) and laboratory information management system (LIMS), the probability of data integrity issues due to human error is decreased but the validation burden and effort to maintain a validated state are higher due to the increased amount of validation testing needed when transferring data from one computer system to another
There have been a large number of issues in regulatory findings regarding data integrity.
Table 2 identifies some of the citations for US FDA Warning Letters and EU Statements of noncompliance for 24 companies [Anonymous, 2015]. Discussion follows. [5]
|
Data Integrity Citations |
Number of Observations in 2014 FDA Warning Letters and EU Statement of Noncompliance for 24 Companies |
|---|---|
|
No adequate controls to prevent unrecorded data changes |
11 |
|
Missing Data |
5 |
|
General Failure to maintain complete and accurate records |
5 |
|
Common Login Information Used by multiple individuals |
5 |
|
Lack of Critical Raw Data |
4 |
|
Altered Files |
4 |
|
System setup to automatically discard negative test results or readings |
2 |
|
Falsified data |
1 |
|
Printing or recording critical information on personal computers |
1 |
|
Lack of validation of computer systems |
1 |
|
Concerns about the quality of clinical trial data |
1 |
|
Electronic records do not meet quality of paper records |
1 |
Table 2: Data Integrity Citations in 2014 for 24 Companies (adapted from Anonymous, 2015)
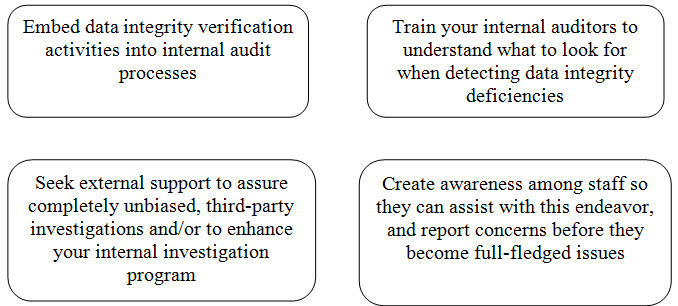
Here are some steps you should take to ensure data integrity:[3]
CONCLUSION
Worldwide Regulatory Agencies does not expect from pharmaceutical industries to review the data by applying Forensic approaches however it is expected that the industries should design the system in such a way that can assure the integrity of data & records with full evidence of supporting raw data that should be secure from any damage, loss & alteration. Due to the increased scrutiny for data integrity, companies are well advised to establish internal competency, assessment and monitoring programs, and assure data integrity is an integral part of the internal audit/self-inspection program.
REFERENCES
1. Jacqueline McCulloch, RAC, Courtney Woodson and Blake Long Data Integrity in the FDA-Regulated Laboratory, regulatoryfocus.org ,April 2014
2. MHRA GMP Data Integrity Definitions and Guidance for Industry March 2015. Medicines and Healthcare Products Regulatory Agency.
3. Siegfried Schmitt, Principal Consultant, PAREXEL® Consulting ‘Data Integrity: FDA and Global Regulatory Guidance’ Jan_2015.
4. www.gov.uk/...data/.../Data_integrity_definitions_ and_ guidance.
5. Jeanne Moldenhauer “Data Integrity Issues in Pharmaceutical Companies: Part 2 | IVT” Jul 16, 2015.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE






